Chapter 10
Humoral Autoimmunity
(Updated 9/13/2012)
L. Yu and G.S. Eisenbarth
Introduction
A little more than 2 decades ago, screening for autoantibodies associated with Type 1A (immune mediated) diabetes was limited to measuring cytoplasmic islet cell autoantibodies (ICAs) and insulin antibodies 1, 2. Currently, multiple sequenced autoantigens have been defined and recombinant autoantibody assays3-7 are available 8, 9 including assays for insulin 10, glutamic acid decarboxylase (GAD) 11, ICA512/IA-2 12-14, I-A2 beta (phogrin) 15, 16, and the islet zinc transporter, ZnT8 autoantibodies 17, 18. Only autoantibody assays to these proteins are of confirmed utility in diagnosing and predicting Type 1A (autoimmune diabetes) and for which laboratories participating in blinded workshops organized by the Immunology of Diabetes Society submit measurements. A long list of additional autoantigens with autoantibody assays exist (see below) but in general we believe given lack of implementation one should assume that these other proposed assays lack sensitivity and or specificity, and the proposed targeted antigen is not sufficiently Type 1A diabetes associated to be of diagnostic utility. In addition to identifying the appropriate target molecules as the basis for Type 1A diabetes autoantibody assays, the assay format is also crucial. Fluid phase radioassays have historically provided the best sensitivity and specificity but this may be changing. Standard ELISA assays where one simply binds a given autoantigen to a plate and detects binding of autoantibodies to the plate bound antigen in general have lacked requisite specificity when large numbers of samples are analyzed. A modified capture ELISA format where autoantibody cross-links a plate bound antigen to the same labeled fluid phase antigen for GAD65, IA-2, and ZnT8 have approached the specificity and sensitivity of fluid phase radioassays. In addition two additional formats (luciferase based assays19 and electrochemiluminescent20) are being studied. The electrochemiluminescent assay format has been applied to the insulin autoantibody which is the most difficult for laboratories to implement 20, 21 with a marked increase in sensitivity above that of even radioassays as evaluated in the last two international Immunology of Diabetes workshops (DASP and IASP).
Just amongst the four major islet autoantibody assays, insulin, GAD65, IA-2, and ZnT8 there are major differences. Insulin autoantibodies are almost always the first autoantibody to appear in children followed from birth (either as an isolated autoantibody or with other autoantibodies)22. The levels of insulin autoantibodies (and only insulin autoantibodies) correlate with the rate of progression to onset of overt diabetes from the time of first appearance of an islet autoantibody6, 23. Thus the youngest onset children who must progress rapidly to be onset at a young age, at onset characteristically have very high levels of insulin autoantibodies. The levels of insulin autoantibodies are not simply related to age, as in general the levels are very low for the whole prediabetic period in young autoantibody positive children who take a long time (e.g. decade) to progress to diabetes(Figure 10.1 below). A recent report suggests that the prevalence and levels of GAD and IA-2 autoantibodies at diabetes onset have increased between 1985 and 200224.
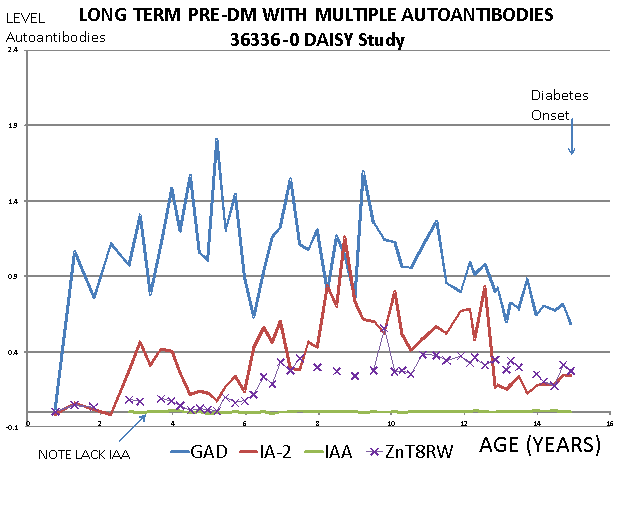
Figure 10.1. Child followed from birth in DAISY study till
development of overt diabetes.
Autoantibodies were present prior to two years of age with more than a
dozen years till onset of hyperglycemia.
Characteristically for children who slowly progress to overt diabetes is
the lack or low levels of insulin autoantibodies.
The most specific islet autoantibodies react with IA-2 and ZnT8 18, 25 and for these two autoantibodies there is characteristically a very strong signal to noise. GAD65 autoantibodies are the most common amongst adult onset patients, but GAD65 autoantibody assays frequently have a 3-5% positivity rate in controls 26 making definitive diagnosis of Type 1A diabetes more difficult if GAD is the only autoantibody present (e.g. diagnosis of Latent Autoimmune Diabetes of Adults) if only GAD65 autoantibodies are present. Though insulin autoantibodies are present in almost all children followed to diabetes, they are frequently negative at onset of diabetes in teenagers and adults.
Expression of multiple autoantibodies of insulin, GAD, IA-2 and/or ZnT8 is associated with extreme risk of progressing to overt Type 1A (immune mediated) diabetes25, 27, 28. Given 20 years of follow up almost all children (whether relatives of patients with Type 1 diabetes or individuals from the general population) who express multiple biochemical autoantibodies progress to diabetes. If there is only a single autoantibody, which are usually insulin autoantibodies or GAD autoantibodies risk of diabetes is low, often less than 5%25, 29. As the number of autoantibodies increase the overall risk of diabetes increases29. The usual explanation for the general rule relating multiple autoantibodies to extreme risk is that epitope spreading with the targeting of different autoantigens directly enhances beta cell destruction. Though insulin autoantibodies often appear first in prediabetic children followed from birth, the order of autoantibody appearance is very variable. After initial appearance with either multiple or a single autoantibody relatively rapidly multiple autoantibodies are present which persist till the development of diabetes. It is doubtful that there is one to one correlation between autoantibodies and T cell targeting of specific islet antigens. In particular with only the level of insulin autoantibodies related to the rate of progression, subjects with low or negative insulin autoantibodies, individuals may still be critically targeting insulin at a low level consistent with delayed progression. Rate of the process needs to be distinguished from risk and it is easiest to do this by only analyzing individuals who have all progressed to overt diabetes with prospective follow up. A life table mixing individual with and without progression to diabetes reflects both rate and risk23.
An alternative hypothesis for the >=2 autoantibody rule, given that Type 1 diabetes related autoimmunity is relatively rare (e.g. 1/300 in general population developing diabetes) is statistical. By Baye’s theorem if one is measuring four autoantibodies (each assay with 1% false positive rate [which only the best international assays achieve in Immunology of Diabetes Society workshops]) the great majority of positives in general population screening are likely to be “false” positives in terms of development of diabetes. A simple way to increase testing specificity is to utilize multiple biochemical autoantibody assays with definition of positive as the presence of >=2 of the four autoantibodies (binomial equations). The combinatorial approach rather than simply asking for all four autoantibodies to be present (four autoantibodies present predicted specificity =(.01)4 or >99.99999%) allows one to preserve sensitivity with a decreased but impressive specificity27. False positives in the general sense for islet autoantibody results can result from assay problems (e.g. we have observed an individual with autoantibodies reacting only with iodinated insulin which does not exist in nature but I-125 iodinated insulin is utilized for the insulin autoantibody radioassay). False positives can also result from true autoantibodies which have low positive predictive values (e.g. “biologic” false positives, low affinity insulin autoantibodies20, 22, ELISA assays for insulin autoantibodies not detecting diabetes related autoantibodies10, etc.). Finally false positives may result from non-antibody serum components that bind or precipitate labeled autoantigens.
Once an individual is identified with islet autoantibodies, follow up for deterioration of glycemic control is important as it is clear from multiple prospective studies that ketoacidosis with both its attendant morbidity and mortality is preventable30 especially in children less than age 2.
In addition to the highlighted major islet autoantigens, there are many other autoantigens in a variety of stages of characterization, including molecules with characterized sequences but without fully developed assays and proteins with only known molecular weights of 155 kd 31, 32, 52 kd 33, and molecules recognized by T lymphocytes for which autoantibodies have not been described such as chromagranin A and IGRP 34, 35 and RegII 36. Other molecules have been described but their association with Type 1A diabetes has either not been evaluated in man, has not been substantiated, or assay sensitivities are low, or follow-up studies have not been published. These molecules include carboxypeptidase H, anti-bovine serum albumin antibodies (anti-BSA) 37, antibodies reacting with ICA69 38-40, anti-insulin receptor antibodies 41, antibodies to heat shock proteins 42-44, anti-topoisomerase II 45 anti-ganglioside 46, lysophospholipids 47and GLIMA38 a membrane glycoprotein 48and antibodies to a series of autoantigens identified by screening islet libraries such as ICA12 49, 50. Finally, a subset of autoantibodies termed anti-islet cell surface antibodies are currently rarely measured as most recent studies have failed to demonstrate disease specificity 51. Recently described autoantigens include osteopontin 52, importin 53, antibodies reacting with “peri-islet Schwann cells/ GFAP/S100beta (glial fibrillary acidic protein)” surrounding islets 54, densin and filtrin 55 and antibodies reacting with CD38 56. There are almost certainly additional specificities awaiting discovery.
Table 10.1: Subset of
Biochemically Characterized Autoantigens
|
|
Sensitivity |
Comment |
|
Insulin |
49-92% |
Higher sensitivity young children/rapid progressors |
|
GAD (Glutamic acid Decarboxylase) |
65-75% |
Higher sensitivity adult onset type 1A |
|
ZnT8 |
65-75% |
Islet Zinc Transporter |
|
ICA512/IA-2 |
74% |
Tyrosine Phosphatase like molecule |
|
IA-2β/Phogrin |
61% |
Tyrosine Phosphatase like molecule |
|
Carboxypeptidase H |
10% |
Infrequent |
|
GLIMA38 |
19% |
amphiphilic membrane glycoprotein |
Despite the importance of autoantibodies for disease prediction 28, it is likely that anti-islet autoantibodies do not by themselves cause the beta cell destruction that leads to Type 1A diabetes. Nevertheless B lymphocytes are important for the development of Type 1 diabetes and results of the TrialNet studies of anti-B cell antibodies (anti-CD20) in new onset diabetes demonstrated slowing of loss of C-peptide. In the NOD mouse, anti-CD20 antibody treatment decreases development of diabetes 57. In addition, Naji and coworkers have demonstrated the importance of transplacental autoantibodies for progression to diabetes of NOD mice 58.
The most cogent evidence of lack of direct damage by
autoantibodies is the lack of diabetes in infants born to antibody positive
mothers, or women who developed Type 1A diabetes during pregnancy.
Autoantibodies (
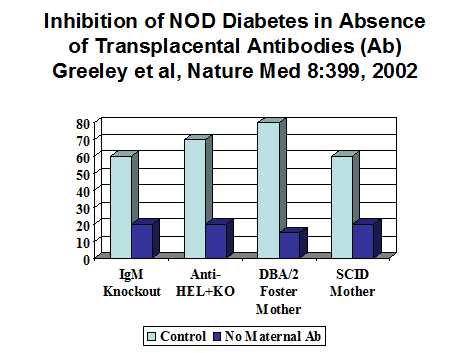
Figure 10.2
Development of diabetes is greatly reduced if maternal autoantibodies are not
present during pregnancy, presumably due to a lack of transplacental
autoantibodies. HEL=Transgenic producing antibodies to Hen Egg Lysozyme; KO=IgM
knockout; DBA/2 strain of mouse used as foster mother; SCID=Severe Combined
Immunodeficient Mother
General Assay
Methodology
There are multiple assay formats for the detection of disease associated autoantibodies, and similar to many fields of autoimmunity, the first useful assay for islet autoantibodies utilized indirect immunofluorescence with frozen sections of human pancreas as substrate (the ICA assay-see below) 1. The other major format for determination of islet autoantibodies consists of variations on the theme of fluid phase assays including recently described luciferase immunoprecipitation assays 19 and electrochemiluminescence assays where the autoantibodies couple a biotinylated autoantigen and fluorophore labeled autoantigen20. In fact the methodology used for the discovery of the radioassay by Berson and Yalow, utilizing insulin antibodies produced by patients treated with insulin, was the basis for the discovery of insulin autoantibodies 2, 64. For most autoimmune disorders and for basic immunologic research, ELISA assays employing plate bound antigen are the standard. Multiple workshops utilizing sera from patients with type 1 diabetes and multiple mouse strains have demonstrated that standard ELISA formats lack both sensitivity and specificity compared to fluid phase radioassay formats. For insulin autoantibodies, the ELISA formats were able to detect high capacity antibodies following insulin immunization, but not the insulin autoantibodies of prediabetic individuals 10. This inability of human insulin autoantibodies to bind to plate bound insulin is specific to human autoantibodies as the insulin autoantibodies of NOD mice can readily be detected in a plate bound ELISA format and we have described a highly sensitive and specific assay for such autoantibodies, equivalent to radioassays 65. We believe insulin bound to plastic plates obscures the limited key determinant(s) recognized by human anti-insulin autoantibodies.
In addition when one analyzes thousands of individuals with ELISA formats there is always a subset of sera that react with the wells of the plate itself or unique epitopes created by plate bound proteins. A small percentage of such sera (e.g. 1%) in studies such as DAISY (Diabetes Autoimmunity Study of the Young) where individuals are sampled on multiple occasions over time, would result in enough false positives to invalidate efforts to discover environmental, immunogenetic, and diabetes predictive parameters 22, 66. It is possible to develop modified ELISA like assays that utilize fluid phase reactivity of antigen and antibody(see discussion of method below). In international workshops an assay for GAD65 autoantibodies (company RSR developed assay and distributed by Kronus) has attained a level of specificity and sensitivity equivalent to the radioassays, though in the most recent 2009 DASP workshop multiple laboratories using this modified ELISA had excellent sensitivity but only 95% specificity (Mueller oral communication). For many applications, 5% false positives would be problematic.
A disadvantage of the current fluid phase radioassays is that they utilize low levels of radiation (usually 125I, 35S -methionine, and 3H-leucine)(Figure 10.3). They are however often performed in a format in 96-well format that is as convenient as the ELISA format 67-69. It is likely that many of the clinical assays (but not all, e.g. transglutaminase) that utilize ELISA formats for other autoimmune disorders are utilizing assays with compromised specificity and sensitivity, and the assay format development has just not been optimized and directly compared with modern fluid phase radioassays (e.g. Farr assay versus, protein A based assay, versus ELISA assay for anti-DNA autoantibodies) 70.
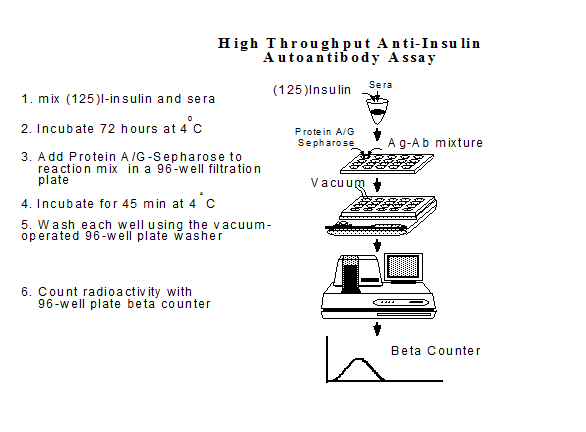
Figure 10.3
General outline of fluid phase 96-well plate assays for
autoantibodies. In above example 125I -insulin utilized, but assay
format identical for GAD65, ICA512 assays.
Most investigators assay GAD65 and IA-2 anti-islet autoantibodies in a high throughput 96-well format (Figure 10.3)where labeled antigen is incubated with patient sera, and then both are placed in 96-well filtration plates, where a "bead" (e.g. sepharose) with coupled protein A and or protein G is added, and bound from free radioactivity is separated by filtration washing, and then scintillation fluid is added directly to the 96-well filtration plates, and counting is performed on multichannel beta counters able to handle the plates 58. Even the assay for insulin autoantibodies utilizing 125I insulin is performed in the same manner, detecting with beta counting emission from 125I (Figure 10.3). A major advance was the simple production of labeled autoantigen by in vitro transcription and translation of cDNAs to produce the label for the fluid phase radioassays. For example, we have utilized a combined GAD65 and ICA512bdc radioassay in which GAD65 is labeled with 3H-leucine and ICA512(IA-2) is labeled with 35S -methionine. It has been our experience in setting up multiple such autoantibody assays that approximately 2/3 of the time an assay using such in vitro labeled autoantigen works, and if the assay does not work on the first try, it is unlikely to be modified to work. At present we routinely determine GAD65, ICA512bdc, IA-2ic, IA-2 full length, ZnT8, 21-hydroxylase and transglutaminase autoantibodies with this methodology 71-75. Such assays are not useful if post-translational modifications, or particular folding of the protein is essential that is not reproduced in the in vitro production of the antigen. With this in vitro translation and transcription methodology minimal protein preparation is needed and following the kit generation of the labeled product we simply perform size separation to produce the labeled antigen. Two harmonized NIDDK assay have been created (for GAD65 and IA-2 autoantibodies) which depended in part on using the same standard sera as well as identical reagents. 76
There are a number of modifications proposed and implemented for the determination of defined islet autoantibodies that can detect autoantibodies with various degrees of sensitivity and specificity relative to the best standard fluid phase radioassays 77-80. In particular the GAD65 autoantibody described by Smith and coworkers 79 utilizes a novel ELISA format in which a low concentration of the GAD antigen on the plate captures the autoantibody, and then biotinylated GAD in the fluid phase is added and is captured by the second binding site of the autoantibody, and it is the biotinylated GAD65 that is detected to produce the non-isotopic signal. This assay performed well in the previous Immunology of Diabetes/CDC DASP workshop, but a similar format for IA-2 autoantibodies did not have an equivalent sensitivity to the standard fluid phase radioassays(oral discussion at IDS-Cambridge and in current 2009 DASP specificity for multiple ELISA assays was only approximately 95%). There is also now a similar ZnT8 commercial assay kit. Human insulin autoantibodies can be measured with an electrochemiluminescent (ECL) insulin/proinsulin assay20. A reported ECL insulin autoantibody assay detected antibodies of NOD mice despite binding of insulin to plate81. I believe it is unlikely for this assay to detect prediabetic insulin autoantibodies in that even though NOD insulin autoantibodies react with plate bound insulin, prediabetic IAA do not 10. There is a need for "point of service" anti-islet autoantibody assays as at present a major portion of the expense of screening for anti-islet autoantibodies relates to handling of the serum specimen and communication with families. We have initial experience with electrochemiluminescent assays for insulin, GAD6S and IA-2 autoantibodies that utilize plate capture and ruthenium labeling of autoantigen which has potential for development of non-radioactive assays20. Screening at the point of service(e.g. primary care office), with follow up laboratory confirmatory assays would enhance the ability to test large populations in a Preventive Health mode, that will likely be necessary if therapies are developed to safely prevent type 1A diabetes.
Islet Cell
Autoantibodies (ICAs)
Studies by groups associated with Blizzard, Nerup, and
In addition to utility in predicting type 1A diabetes, islet autoantibodies aid in the diagnosis of type 1A diabetes. Approximately 7% of patients with initially non-insulin dependent adult onset diabetes have GAD autoantibodies 87-89. These GAD positive individuals are at increased risk to become insulin dependent 87(though multiple autoantibodies likely superior prediction) 90 and they are referred to as having LADA (Latent Autoimmune Diabetes of Youth) diabetes 87, 91. It is likely that for patients with typical Type 2 diabetes (e.g. > age 45, obese) presence of a single islet autoantibody depending on the assay specificity may be a “false positive” (e.g. 4 antibodies with 99% specificity, 4% false positive; 4 antibodies 97.5% specificity, 10% false positive) and presence of ≥ 2 islet autoantibodies may be required for diagnosis of Type IA diabetes. Of note, measurements of ZnT8 autoantibodies have recently been reported to be of utility in characterizing adult onset diabetes 92. For children and adults93 clinically diagnosed with Type 2 diabetes, presence of islet autoantibodies as expected are associated with insulin deficiency 94.
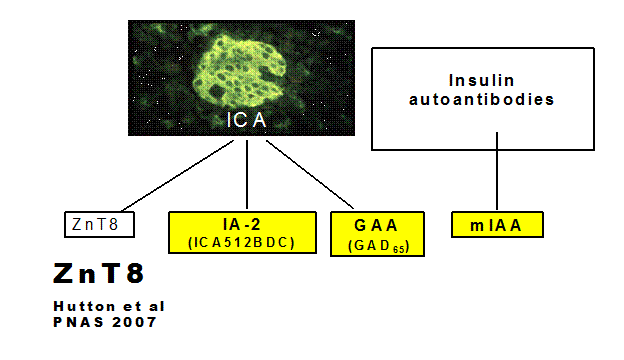
Figure 10.4: GAA (GAD
autoantibodies) ZnT8 and IA-2 autoantibodies contribute to ICA reactivity, but
insulin autoantibodies do not.
Data from the large DPT-1 (Diabetes Prevention Trial) study demonstrated that ICA autoantibodies, as well as GAD autoantibodies and IA-2 autoantibodies, in relatives were relatively stable with 75-85% of all positive patients (90 to 95% with high levels) confirmed during follow up. Approximately 5% of DPT-1 patients without biochemical autoantibodies did progress to diabetes, emphasizing the likely presence of additional autoantigens including ZnT8 autoantibodies which were not measured25.
Since the discovery of cytoplasmic
Limits of the
To date, three autoantigens have been defined that
contribute to the ICA positivity (Table 10.1, Figure 10.4). The
best-characterized subset of
Other defined antigens accounting for a subset of
The standard "biochemical" autoantibody assays measure antibodies reacting with insulin, GAD65, IA-2 (also termed ICA512) and ZnT8. It has been much easier to standardize assays for GAD65 and IA-2 as compared to insulin autoantibodies 109 which probably relates to the much stronger signal/noise for GAD65 and IA-2 autoantibodies 110. Receiver Operator Curves demonstrate a very small difference in signal between prediabetic/diabetics and the bulk of normal control sera for insulin autoantibodies. Thus this assay requires meticulous attention to detail, in contrast to GAD65 and IA-2 assays that are more "forgiving". With the Immunology of Diabetes Society (IDS) and Centers for Disease Control DASP workshop program standard sera are now available and help with assay implementation (see http://www.immunologyofdiabetessociety.com/).
Sequenced
Autoantigens
Glutamic Acid
Decarboxylase (GAD)
The discovery that anti-GAD autoantibodies accounted for an
important component of anti-64 kd autoantibodies and a subset of
Several different radioassays for anti-GAD autoantibodies have been developed and multiple Immunology of Diabetes Workshops (IDW, now IDS (Immunology of Diabetes Society)) for measuring such antibodies were held 109. Four early formats for determining anti-GAD autoantibodies include: (1) determination of antibody precipitation of GAD enzymatic activity, (2) radioassays utilizing affinity purified porcine brain GAD that has been labeled with 125I, (3) radioassays utilizing endogenously labeled GAD produced by in vitro transcription and translation of GAD cDNA 121, and (4) ELISA assay formats. These assay formats are likely to give very different results depending upon the population studied. In particular, the immunoenzymatic assay appears to be less sensitive than either radioassay. Radioassays utilizing in vitro transcribed and translated GAD65 or 125I -labelled recombinant human GAD65 can give equivalent results with high sensitivity and specificity. The Combinatorial Autoantibody International workshop of the Immunology of Diabetes Society indicated that most laboratories (but not all) concordantly measured anti-GAD65 autoantibodies 109. All attempts at standard ELISA assays have failed to achieve the sensitivity and specificity exhibited by most of the fluid phase radioassays. Recently, a modified ELISA assay where a low concentration of GAD65 bound to plates is recognized by one “arm” of anti-GAD autoantibodies and then the other arm captures biontinylated GAD65 to produce a non-radioactive assay for GAD65 autoantibodies 79. In addition, luciferase based (LIPS: Luciferase based autoantibody Assays)19, 122 liquid-phase luminescence immunoprecipitation system and electrochemiluminescent assays are being applied to multiple autoantibody assays.20
The Immunology of Diabetes Society (IDS) and Centers for Disease Control (CDC) have organized multiple DASP workshops. The majority of participating laboratories utilize fluid phase radioassays. A standard calibration sample (World Health Organization) was distributed and used in these workshops and results for GAD65 and IA-2 autoantibodies can be reported in WHO units, or as an index based upon a standards reactivity. Despite the reporting of GAD autoantibodies in WHO units, the utilization by each laboratory of their own standard samples, quantitative concordance between laboratories can be limited, and it is likely to obtain better correlation the same standard sera might need to be utilized in each laboratory. This is not possible except for limited consortia (e.g. NIDDK Harmonization76), and thus though reported levels of autoantibodies correlate, exact levels reported by different laboratories differ.
The quantitation of anti-GAD autoantibodies can aid in the
detection of extremely high titers of such antibodies associated with restricted
Among ICA-positive first-degree relatives, the levels of anti-GAD autoantibodies are remarkably constant with up to a decade of prospective evaluation. Harrison and coworkers 123 have reported that there is an inverse correlation between levels of GAD autoantibodies and a T cell proliferative response to GAD. Thus it is hypothesized that high levels of GAD autoantibodies may be associated with a diminished anti-islet T cell response (Th 1 rather than Th2) and therefore less or no beta cell destruction. Given perhaps the difficulties of T cell assays this observation has not been confirmed and in general dividing islet autoantibodies into quartiles, the higher the level of the islet autoantibody the greater the risk of progression to diabetes for at risk relatives 107, 124.
Two isomers of GAD, GAD65 and GAD67, share an identical exon-intron structure and are 76% identical in amino-acid sequences with only 22% identity in N-terminal region and over 90% identity in C-terminal regions 125. Antibodies to GAD65 were detected in over 80% of the patients with type 1A diabetes 27 versus only 11-18% having antibodies to GAD67 126, 127. The autoantibodies binding to GAD67 in IDDM seem to represent antibodies to shared epitopes with GAD65 126 although there was a report that the patients with Grave's disease have been found carrying specific GAD67 antibodies in the absence of GAD65 antibodies 128.
GAD67 and GAD65 have similar overall structure that is important for antibody binding. GAD65/GAD67 chimeric proteins were widely utilized in most of laboratories to define the epitope of GAD65 while maintaining the overall GAD protein conformation. The precise mapping of GAD epitopes targeted by patients with type 1A diabetes is difficult because serum contains polyclonal antibodies with multiple specificities. Monoclonal antibodies derived from individuals with new-onset diabetes have been useful in mapping GAD epitopes in type 1A diabetes 129-131. Two major epitope regions targeted by human diabetic sera have been identified 132 and confirmed by other laboratories. One epitope is in the middle part of GAD65 between amino acids 240 - 435 (termed IDDM-E1) and one is at the C-terminal region of GAD65 between amino acids 451 - 570 (termed IDDM-E2). Baekkeskov, et al 133 used homolog-scanning mutagenesis to identify GAD65-specific amino acid residues which form autoreactive antibody epitopes in this molecule. Detailed mapping of 13 conformational epitopes, recognized by human monoclonal antibodies derived from patients, together with two and three dimensional structure predictions led to a model of the GAD65 dimer. The results revealed a remarkable spectrum of human autoreactivity to GAD65, targeting almost the entire surface of the molecule and reactivity to the multiple epitopes are present both before and after onset of type 1A diabetes 134, 135. This suggests that native folded GAD65 is the immunogen for autoreactive B-cells in IDDM.
Comparisons of the autoimmune repertoires to GAD65 in type 1 diabetes, Stiff Person Syndrome, and Autoimmune Polyendocrine Syndrome (APS) might provide insight into why some individuals develop one disease and other individuals develop the other disease. Most type 1 sera only target GAD65 protein epitopes dependent on protein conformation and do not bind GAD65 protein fragments or react with denatured GAD65 protein with immunoblotting 11, 132. In contrast, Stiff Person Syndrome sera have antibodies that bind GAD65 protein fragments consisting of N-terminal, middle, and C-terminal regions 132, 136-138. Characteristically, Stiff Person Syndrome sera target an epitope contained in the N-terminal first eight amino acids and neither IDDM nor APS sera have such antibodies. Despite clear differences in the GAD65 antibody profile in Stiff Person Syndrome and IDDM, an important similarity exists. Both Stiff Person Syndrome and IDDM sera contain antibodies that bind a conformation-dependent region in the middle part of the protein and a second set of antibodies that binds a conformation-dependent epitope in the C-terminal regions of the protein 132. Antibodies specific for these two regions of GAD65 are present in approximately equivalent titers in most sera although a single specificity may predominant. Like Stiff Person Syndrome, APS1 sera have antibodies that inhibit the enzymatic activity of GAD65, while this is not a property of the usual type 1 diabetes autoantibodies 138. The similarity and difference in the GAD65 antibody repertoire in the Autoimmune Polyendocrine Type 2 Syndrome (APS2) and diabetes has been studied 139. Neither diabetes nor APS2 sera bind the N-terminal third of GAD65, but instead target the C-terminal two-thirds of GAD65, IDDM-E1 and IDDM-E2. More detailed mapping has found differences for APS2 sera reactivity in the IDDM-E2 region 139. There are multiple reports of GAD65 autoantibodies associated with interferon alpha therapy, and in particular with therapy for viral hepatitis. Schories and coworkers described de novo development of GAD65 autoantibodies in 2/74 patients treated with interferon-alpha for hepatitis C infection and recommended testing for such autoantibodies prior to a second course of therapy, given the progression to overt diabetes of one of their GAD65 positive patients 140-142.
Hampe and coworkers have developed a novel assay for GAD epitopes utilizing competition with Fab fragments of monoclonal anti-GAD antibodies 143. Their group recently reported the detection of anti-idiotypic antibodies in the sera of normal individuals which they believe mask the presence of GAD autoantibodies in all normal individuals 144. They contend that patients expressing GAD autoantibodies lack these anti-idiotypic antibodies and thus are positive with current GAD assays. This is a very novel suggestion and will require more detailed study145. In particular the assay used to analyze anti-idiotypic antibodies involves incubating serum with a human anti-GAD monoclonal covalently bound to beads and then centrifuging the mixture to remove “anti-idiotypic” antibodies. There is the possibility for the monoclonal anti-GAD antibody to be released from the beads and thus give the appearance of anti-idiotypic antibodies. The claim that positive diabetic sera does not have anti-idiotypic antibodies is primarily that such sera gives signals with or without absorption with the beads 144.
ICA512 (IA-2) and IA-2 β (phogrin)
Following the identification that a major component of the 64 kd antigen is GAD, many groups reported an association between Type 1A diabetes and anti-GAD antibodies. Evidence for heterogeneity of anti-64 kd autoantibodies came from the work of Christie 146, who found that antigenic tryptic fragments were generated after mild trypsinization of 64 kd molecule(s). In particular, precipitation of human islets with sera from nondiabetic polyglandular failure individuals, followed by trypsinization, yielded a 50 kd fragment whereas a 38-40 kd fragment was precipitated by new-onset and prediabetic sera. Of note, mild trypsinization of the GAD molecule yields a 50 kd fragment. Reports 146, 147 indicated that autoantibodies to the 37-38 kd fragment had a higher positive predictive value for diabetes than anti-GAD antibodies. It appeared that there existed two different 64 kd autoantigens in human islets, one being GAD and another two molecules that after trypsinization yield a fragment of 37-40 kd. Christie reported that the 40 kd fragment is related to the protein tyrosine phosphatase IA-2 (ICA512) 148 and the 37Kd fragment to the molecule IA-2 β 16.
The autoantigens ICA512 (IA-2) and subsequently IA-2 β (phogrin) were isolated
independently by several investigators probably reflecting the importance of
these related molecules. Rabin and coworkers 12
screened an islet expression library with sera from patients with type 1A
diabetes. The molecule he isolated was termed ICA512. The same molecule has
been termed IA-2. The original sequence in GenBank from Rabin and coworkers
lacked several nucleotides out of approximately 2,000, but with a frame shift
predicted a shortened protein, which was originally utilized in autoantibody
assays. A related protein with homology in a putative tyrosine phosphatase
region (though to date no enzymatic activity has been demonstrated for either
molecule) was termed IA-2 β
by Notkins and c
There is additional heterogeneity of the autoantibodies
which react with IA-2(ICA512). A large number of epitopes (>10) within the
intracytoplasmic C-terminus of the molecule are targets of autoantibodies 149. In addition islet cells
express differentially spliced messenger RNA for ICA512, with one form lacking
exon 13 which includes the transmembrane region of the molecule. In our
laboratory by screening an islet expression library we obtained an ICA512 clone
(ICA512bdc) which was used to develop an autoantibody radioassay 150, 151.
This clone lacks exon 13 [construct termed ICA512bdc-(BDC for
Two epitopes of particular interest within the ICA512 molecule are a mini-epitope described by Dotta and coworkers at the C-terminus of the molecule 153 and a juxtamembrane epitope described by Bonifacio and coworkers 59 (Figure 10.5). The mini-epitope is of interest in that despite in vitro transcribing and translating only 51 amino acids of the ICA512 molecule, approximately 56% of patients who have ICA512 autoantibodies react with this epitope 154. Bonifacio and coworkers have evidence that autoantibodies to the juxtamembrane region (amino acids) are amongst the first to appear in individuals developing type 1 diabetes. In our studies of young infants we have not been able to confirm this finding but the number of such children we have studied developing antibodies de novo is small.

Figure 10.5 In Vitro
transcription and translation of fragments of alternative splice variants of
ICA512 and smaller fragments recognized by ICA512 autoantibody positive sera.
Insulin
Insulin is a 51-amino acid disulphide-linked heterodimer that is specifically produced by beta cells of islets (Figure 10.6). Proinsulin which is processed within the secretory granule is the precursor to insulin but exactly how it is processed to become an autoantigen is a key question155 with evidence that a selected insulin B:9-23 epitope is created specifically in islet beta cells156-161. To date insulin and proinsulin are the only known beta cell-specific autoantigens within human islets (all other putative autoantigens are produced by human non-beta islet cells). Of note, both in mouse and man, proinsulin messenger RNA and proinsulin are present in the thymus 162-167. Transgenic mice with transgenes with the rat insulin promoter coupled to several proteins indicate that thymic transcription of the insulin gene is possible 168. Insulin in islet beta cells is produced as a preprohormone that is processed to pro-insulin and then, with removal of connecting peptide (C peptide), to mature insulin. Processing occurs within beta cell secretory granules, where insulin is packaged in a crystal form 169. Equimolar concentrations of insulin and C peptide are secreted by beta cells. In the thymus processing is likely to be very different and thus the epitopes presented that delete autoreactive T lymphocytes. Insulin is almost certainly the primary target of autoimmunity of NOD mice and likely man (e.g. insulin gene polymorphism associated with increased diabetes risk associated with decreased thymic insulin message, levels insulin autoantibodies correlating with rate of disease, insulin autoantibodies usually first autoantibody to appear)161, 170.
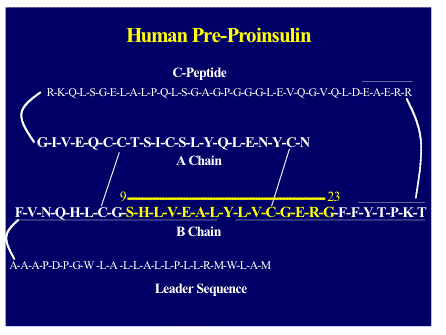
Figure 10.6: Sequence
in pre-proinsulin with the NOD mouse insulin B chain (B:9-23) dominant T cell
epitope highlighted.
In the 1950s, Berson and Yalow developed the first radioimmunoassay utilizing sera with insulin antibodies from patients treated with bovine insulin 171. Bovine insulin differs from human insulin at 3 of 51 amino acid and porcine insulin differs from human insulin by one amino acid (at the terminus of the B chain). For most patients treated with subcutaneous insulin, the presence of anti-insulin antibodies does not interfere with insulin therapy. A subset of insulin-treated patients with extremely high levels of insulin antibodies are insulin resistant with mean insulin binding capacities greater than 216 nM (30,000 microunits of insulin/ml serum) 172. For such patients, the species of insulin used for therapy is usually changed (e.g., human insulin to an analogue insulin or vice versa and occasionally to sulfated insulin) . Anti-insulin antibodies during pregnancy may facilitate transplacental passage of animal insulins.
A very rare, but interesting syndrome is the Insulin Autoimmune Syndrome, also termed Hirata syndrome 173, 174. Patients with this syndrome (almost all with DRB1*0406) after exposure to sulfhydryl containing medications (e.g. methimizole, penicillamine) develop extremely high levels of insulin autoantibodies 175. These patients usually present with hypoglycemia which resolves with discontinuation of the medication. In addition to this MHC restricted syndrome, some patients have monoclonal insulin autoantibodies produced by B lymphocyte tumors 174.
The production of antibodies to animal insulin was not unexpected. It was, however, discovered that individuals treated with human insulin also produce anti-human insulin antibodies (though at a lower concentration) 172. The development of anti-insulin antibodies following subcutaneous therapy with human recombinant insulin has been ascribed to the possible presence of "denatured" insulin molecules. It is, however, very likely that self proteins when administered subcutaneously, especially in a depot form (e.g., complexed with the positively charged protein NPH (neutral protein Hagedorn), or zinc, as in the two most common pharmacologic long-acting insulin preparations), will induce antibodies. Antibodies also develop following therapy within a number of other recombinant proteins including human growth hormone and factor VIII. To test the generality of production of insulin antibodies following subcutaneous insulin administration, we transplanted a pituitary cell line producing rat insulin (which has the same sequence as mouse insulin) into histoincompatible mice. With rejection of the tissue, anti-insulin "autoantibodies" were induced 176.
In 1983 Palmer and coworkers 2 discovered the presence of anti-insulin antibodies in patients with new-onset Type 1A diabetes prior to the administration of exogenous insulin. Subsequently a large number of studies have demonstrated that anti-insulin antibodies are present for years before the development of Type 1A diabetes 177. Initially, two different assay formats were utilized to detect such antibodies (an ELISA format with insulin immobilized on plates and fluid phase radioassays). The two different formats gave very different results relative to the positive predictive value for development of Type 1A diabetes. A series of international workshops and sera exchanges led to the observation that the two formats measure different antibodies, and that only the antibodies detected with the radioassay formats were associated with risk for Type 1A diabetes 10. Both assay formats detected antibodies following insulin therapy and rarely were positive in normal controls. We have evidence that the inability of standard ELISA assays with insulin bound to plates is specific to human insulin autoantibodies as we have developed ELISA format assays that readily detect the insulin autoantibodies of the NOD mouse 65.
As more has been learned concerning insulin autoantibodies, the inability of standard ELISA assays to detect disease-relevant antibodies is understandable and probably relates to obscuring a key epitope of insulin when bound to a solid phase. In particular, the anti-insulin autoantibodies associated with diabetes risk are all of extremely high affinity and, most important, of very low capacity (10 -12 M), making their detection with plate-binding assays problematic 10. The epitope of the insulin molecule recognized with fluid phase assays appears to be homogeneous 178. In particular, the antibodies from all antibody-positive relatives we have studied react with a conformational epitope (do not react with either the A or B chain) and react equally well with insulin and proinsulin 22. To date, all insulin autoantibodies of at-risk relatives react with des B23 to B30 insulin lacking the terminal eight amino acids of the B chain. Des B23 to B30 insulin fails to bind to the insulin receptor. The A13 leucine is in the base of a pocket of the insulin molecule, and if this leucine is replaced by a tryptophan that extends out of the pocket, insulin autoantibody reactivity is abrogated. The reason for this marked specificity of insulin autoantibodies reacting with the face opposite the insulin-binding domain is unknown. It is, however, very likely that the antibody response to insulin is "antigen-driven", giving rise to high-affinity autoantibodies. Insulin may react with B cells while bound to insulin receptors, either of neighboring immunocytes or perhaps following its transport on insulin receptors through the endothelium. The ability to produce insulin analogues that are recognized by insulin autoantibodies but not by the insulin receptor suggests that one can selectively target B cells producing such antibodies. Whether elimination of such B cells which are probably extremely efficient at presenting insulin to T cells, would influence beta cell destruction is unknown. Autoantibodies of prediabetics which react with proinsulin are all absorbed with insulin, and assays for insulin autoantibodies are reported to have higher disease specificity compared to proinsulin autoantibodies. We have recently developed a modified insulin autoantibody ELISA assay for the autoantibodies of the NOD mouse that is as sensitive as our standard radioassay while retaining specificity, but to date the same assay format does not detect human prediabetes anti-insulin autoantibodies 65. A portion of the difficulty we believe relates the marked epitope specificity of human autoantibodies with plate binding interfering with this reaction as well as additional difficulties created by variable background binding of different human sera to plates +/- antigen.
Levels of insulin autoantibodies, similar to GAD autoantibodies, appear to be regulated at different levels over long periods of time in prediabetic first-degree relatives (Figure 10.1). Of interest is a report indicating that in a subset of children who are negative for insulin autoantibodies at onset, insulin autoantibodies are detectable in their IgG serum fraction, suggesting blocking of reactivity by immune complexes 179.
The levels of insulin autoantibodies correlate inversely with the age at which type 1 diabetes develops180. Thus levels greater than 2000 nU/ml are almost exclusively found in patients who progress to Type 1A diabetes prior to age 5, and less than half of individuals developing Type 1A diabetes after age 15 have levels of anti-insulin autoantibodies at diagnosis distinguishable from controls. Of note the levels of insulin autoantibodies are not simply age related, but related to the rate of progression to diabetes from the appearance of islet autoantibodies (See figure 10.1) 23. The levels of such antibodies are to some extent genetically influenced(e.g. insulin gene polymorphism) and are associated with DR4 181 and DQ8 22, and are also associated with other haplotypes with DQA1 alleles of lineage 2, DQA1*0102, 0201, 0301, 0401 181. Presence of anti-insulin autoantibodies and their levels appear to be associated with such lineage 2 alleles in a dominant manner. In particular, individuals developing type 1A diabetes who are homozygous for DQA1*0501/DQB1*0201 (DR3 homozygotes) either are negative for anti-insulin autoantibodies or have very low levels. There is data that the levels of anti-insulin autoantibodies among ICA-positive first-degree relative correlate with the rate at which individuals progress to overt diabetes 180 that we have recently confirmed for children followed from birth23. Relatives, however, who only express anti-insulin autoantibodies infrequently progress to overt diabetes 22. A proportion of anti-insulin autoantibody-positive, ICA-negative relatives under the age of 10 convert to ICA positivity.
Williams and coworkers modified the insulin autoantibody assay by utilizing protein A to precipitate autoantibodies 182 rather than polyethylene glycol used in the initial standard insulin autoantibody assays 2. The assay (termed micro-insulin autoantibody assay) was modified such that rather than 600ul of sera/sample, the assay utilized approximately 25ul of sera per sample for duplicate determination with and without competition with non-labeled insulin. Not only did the assay utilize less sera, it also eliminated two artifacts apparently related to the use of polyethylene glycol (polyethylene glycol precipitates many proteins based on a protein's solubility). Both cord blood and hemolyzed sera have factors, which bind labeled insulin and are precipitated by polyethylene glycol while both types of sera are negative with assays utilizing protein A precipitation 183, 184. We have further modified the Williams assay by utilizing a 96-well plate format with membrane filtration to separate bound from free autoantibodies, and direct counting on a beta counter of the precipitated 125I -insulin. The counting in a 96-well plate, and handling in 96-well plates allows semi-automated determination of insulin autoantibodies in a format similar to that for the GAD65 and ICA512 autoantibody assays. A recently developed electrochemiluminescent assay for insulin autoantibodies appears to be more sensitive than current radioassays and has the advantage of not requiring radiation(Figure 10.7)20.
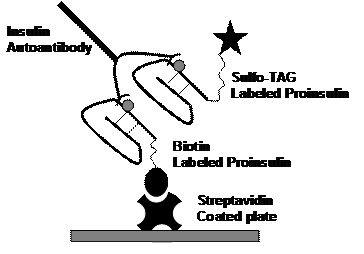
Figure 10.7: Electrochemiluminescent
Assay for insulin autoantibodies. Yu et
al Diabetes 61:179-186, 2012
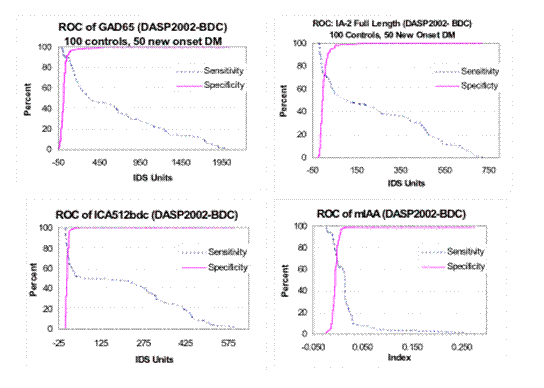
Figure 10.8: ROC
curves of autoantibodies, Denver Laboratory DASP Radioassays
Unlike GAD65 and IA-2 autoantibody assays, mIAA assays are poorly correlated between laboratories of the DASP Immunology of Diabetes/CDC workshops including the 2009 DASP workshop (Mueller oral presentation). In the DASP workshops to date, assay sensitivities have been relatively poor for the majority of laboratories (<30%), with only a handful of laboratories having sensitivities (with preserved specificity) exceeding 50%. The sera samples of the patients with diabetes in the DASP workshops are predominantly obtained from older individuals with type 1A diabetes given the volumes needed for the workshops. Since subjects with diabetes onset as adolescents and adults have markedly lower levels of insulin autoantibodies compared to younger children, difficulty with measuring these low-titer anti-insulin autoantibodies is not surprising. As illustrated (Figure 10.8) by the ROC curves above, the signal to noise ratio for insulin autoantibodies (mIAA: micro-insulin autoantibodies) of DASP workshop samples is very small (lower right panel) compared to the curves for GAD65, full length IA-2 and ICA512bdc (IA-2 variant lacking exon (transmembrane) 13).
Insulin autoantibodies are usually the first autoantibody to
appear in young children developing type 1 diabetes 22, 185, 186. This is particularly
true for infants less than 1 year of age who begin to express
autoantibodies. Achenbach and coworkers
have analyzed the affinity of anti-insulin autoantibodies for children followed
prospectively in the BabyDiab study (Figure 10.9). As illustrated in the figure
below a high percentage of the children who went on to develop multiple
anti-islet autoantibodies or to progress to diabetes express high affinity
autoantibodies (>10(9) l/mol). In addition the high risk, high affinity
autoantibodies differed from the autoantibodies of children who failed to
develop additional autoantibodies (remained IAA positive only) or had transient
insulin autoantibodies in that the majority reacted well with proinsulin 22. It is likely that most
of the lower affinity proinsulin non-reactive insulin autoantibodies are
"false positive" autoantibodies relative to diabetes risk. In a study by Siljander from Finland, he did
not find differential affinities of insulin autoantibodies but it is not clear
if the insulin autoantibody assay was of similar sensitivity to that of
Achenbach’s study 187 To aid in screening for the affinity of
insulin autoantibodies Curnock et al have reported utilization of a single
concentration of unlabelled insulin as competitor24.
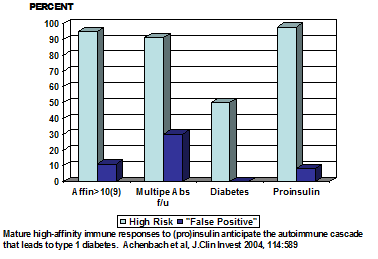
Figure 10.9: Insulin
autoantibodies of BabyDiab children.
Isotypes of insulin autoantibodies have been evaluated in
the BabyDiab study and in studies from
In addition the micro-IAA assay readily detects insulin autoantibodies in NOD mice and the early expression of insulin autoantibodies of NOD mice correlates with the early development of type 1 diabetes. Approximately 90% of NOD mice expressing insulin autoantibodies at 8 weeks of age develop diabetes by 16 weeks of age. Figure 10.10. Insulin autoantibodies can be rapidly induced in normal Balb/c mice with the administration of the autoantigenic peptide, residues B:9-23 of insulin. These antibodies do not react with the immunizing peptide but with intact insulin 189. This surprising finding clearly indicates that normal mice have autoreactive T and B cells able to respond to the peptide (T cell) and to intact insulin (B cell). Further studies using the B:9-23 peptide and poly-IC (viral mimic and activator of the innate immune system) in Balb/c mice have generated insulitis and in Balb/c mice with transgene induced B7.1 islet expression, diabetes is induced. Thus immunization with this single peptide can lead to the destruction of islet beta cells 190-194
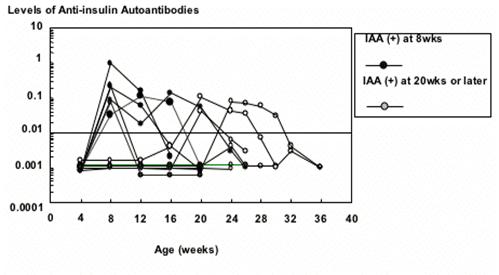
Figure 10.10:
Development of insulin autoantibodies in individual NOD mice. Mice which
expressed insulin autoantibodies at 8 weeks of age developed diabetes prior to
16 weeks, while mice expressing insulin autoantibodies after 20 weeks developed
diabetes later. Yu et al. PNAS 97:1701, 2000.
Two workshops have evaluated insulin, GAD65, and ICA512 (IA-2) autoantibodies in NOD mice. The workshop reports conclude that insulin autoantibodies measured by a sensitive radioassay are strongly associated with autoimmunity while neither GAD65 nor ICA512 (IA-2) specific autoantibodies were demonstrated 195.
Thomas and coworkers have produced a series of monoclonal insulin autoantibodies and recently transgenic mice producing an insulin autoantibody 196. These mice are tolerant to insulin, suggesting that even low levels of insulin can induce B cell tolerance. Proinsulin is present in thymus and lymph node. Knocking out of the insulin 2 gene (the insulin of mice expressed within the thymus as well as islets) greatly accelerates the development of diabetes and increases levels of insulin autoantibodies 161, 190, 197, 198. In contrast an insulin 1 gene knockout on the NOD background prevents the development of diabetes in the majority of mice but has relatively little effect upon insulin autoantibodies 190. Knocking out both the insulin 1 and the insulin 2 gene and replacing insulin with a mutated preproinsulin gene (B16:A rather than B16:Y) completely prevents the development of NOD diabetes and greatly diminishes insulin autoantibodies 161, 199. We believe the insulin peptide B:9-23 is a primary autoantigenic determinant of the NOD mouse. This peptide is presented to diabetogenic T cell receptors in a low-affinity register bonding to MHC Class II I-Ag7 and binding in a low affinity register may relate to escape of anti-insulin B:9-23 T cells from thymic deletion despite expression of low amounts of insulin in the thymus 158.
ZnT8
The fourth major confirmed islet autoantigen recognized by human autoantibodies is the islet specific zinc transporter ZnT85, 25, 200-202. It was discovered to be an autoantigen by Hutton and coworkers based on algorithms searching for beta cell specific highly expressed molecules 17. The ZnT8 transporter is one of a large family of zinc transporters and ZnT8 is associated with the membrane of secretory granules of islet beta cells. Zinc within secretory granules of beta cells is complexed with insulin, forming a storage crystal composed of zinc and insulin.
An initial fluid phase radioassay for anti-ZnT8 autoantibodies utilizing the full length molecule had a relatively low sensitivity. Most of the full length molecule is hydrophobic as it is within the membrane of secretory granules. When Hutton and coworkers developed assays utilizing the C-terminus of the molecule highly specific and sensitive assays were achieved17, 18, 25, 202-207. Autoantibodies reacting with ZnT8 are present in the majority of patients with Type 1 diabetes and assay specificity and sensitivity is similar to those for GAD65 autoantibodies in DASP workshops. Of note there are two major polymorphic variants of ZnT8, one with a tryptophan and the other an arginine at position 325 of the molecule 204 (also a Q variant is more common in African Americans). The majority of patients have autoantibodies recognizing both variants but a subset have autoantibodies recognizing only one variant. Those recognizing the specific arginine variant are patients genetically homozygous for the arginine variant, and vice versa for the tryptophan variant. This is a unique demonstration that islet autoimmunity in terms of epitopes recognized is truly autoimmune, with patients recognizing their own sequence. If ZnT8 were discovered prior to GAD65 or IA-2 autoantibodies it would be part of the primary panel of autoantibodies. At present we measure ZnT8 autoantibodies in diabetic patients negative for the other biochemical autoantibodies in whom we attempting to diagnose Type 1A diabetes. For at risk populations such as relatives of patients with Type 1 diabetes and individuals with high risk HLA alleles from the general population we measure ZnT8 autoantibodies in those expressing a single anti-islet autoantibody. If ZnT8 autoantibodies are present these individuals would have >=2 anti-islet autoantibodies and have a higher risk of progressing to diabetes 17. In general, ZnT8 are often the last autoantibody to appear in children followed from birth and often after onset of diabetes become negative the soonest 206 208.
ICA69
Pietropaolo and coworkers identified a novel islet protein termed ICA69 38 through the screening of a lambda gt11 human islet expression library with ICA-positive sera 209. This protein, which on SDS gel migrates at 69 kd, has been sequenced and found to have in molecular weight of 54,600 (the aberrant gel migration is probably explained by the presence of several highly charged region in the molecule, as ICA 69 is not glycosylated). ICA69 is present, at least at the levels of messenger RNA, in brain, lung, kidney, and heart, with high levels in islet and other neurendocrine tissues. Pietropaolo and coworkers have reported that ICA69 expression in the thymus of NOD mice is reduced 210. In the rat, ICA69 is beta cell specific 211. ICA69 by confocal microscopy is associated with the Golgi-complex and immature (lesser extent) insulin secretory granules with essentially no ICA69 evident on secretory granules near the plasma membrane 40. A knockout of ICA69 in C. elegans compromised neurotransmission 212. A knockout of the ICA69 gene (ica-1) was bred onto the NOD mouse and insulitis and diabetes developed normally in these knockout mice 213.
ICA69 is identical in sequence to the cow's milk-related protein p69 described by Dosch and coworkers 214. It has been reported that diabetic individuals have antibodies to bovine serum albumin and there are a number of epidemiological studies indicating that the neonatal ingestion of cow's milk increases the development of Type 1A diabetes. ICA69 and bovine serum albumin have two short regions (five amino acids) of identity and in the region of the ABBOS albumin peptide (potential T cell epitope), four of nine amino acids are identical. Such identity may be enough to stimulate cross-reactive T cell clones.
In the initial assay format, antibodies to ICA69 were measured by Western blot utilizing recombinant ICA69. The Western blot assay format relative to radioassays for antibodies reacting with insulin, GAD65, and ICA512 is inadequate, with more than 5% of normal controls reacting on Western blots. Anti-ICA69 autoantibodies are diabetes-related autoantibodies, but are present also in rheumatoid arthritis patients 215. Further studies are needed to elucidate the role of this molecule in the pathogenesis of Type 1A diabetes. A report by Atkinson and coworkers questions the association with Type 1A diabetes of anti-albumin antibodies and of T cell responses to albumin 216. Dosch and coworkers however in studies of mice can accelerate or inhibit development of diabetes with a peptide of the mouse molecule 217.
Carboxypeptidase H
Carboxypeptidase H is another autoantigen discovered by the screening of islet expression libraries with prediabetic sera. Within islets, carboxypeptidase H cleaves carboxyterminal amino acids during the processing of proinsulin to insulin. This molecule is not islet specific, being present also in bovine adrenal, pituitary, and kidney 169. In beta cells, carboxypeptidase H is localized in the insulin secretory granule, where it exists both in a membrane bound (52 kd) and soluble form (50 kd). Carboxypeptidase H is probably the most abundant islet protein after proinsulin-insulin. A radioassay or an ELISA for anticarboxypeptidase antibodies has not been developed. In the original assay format utilizing reactivity with recombinant Escherichia coli plaques, one of four prediabetics had anti-carboxypeptidase H antibodies.
ICA12
In the screening of islet expression libraries with patient autoantibodies Rabin and coworkers identified a molecule they termed ICA12 12 in addition to ICA512, discussed above. The complete sequence of ICA12 is now known and radioassays have been developed with in vitro transcription and translation of ICA12. ICA12 is the transcription factor SOX13. Only a small percentage of patients with new onset diabetes express ICA12 autoantibodies 49. It is not clear whether the percentage of patient’s positive for ICA12 autoantibodies, compared to controls with other autoimmune disorders, is high enough for ICA12 autoantibodies to be truly type 1 diabetes related, including increased levels in primary biliary cirrhosis 218, 219.
Partially Characterized Autoantigens
52 kd
A 52 kd antigen distinct from carboxypeptidase has been identified by Western blotting of human islet extracts with diabetic sera 33, 220. Interestingly, this antigen that appears to be islet cell-specific shares an antigenic determinant with the protein PC2, a component of the rubella virus capsid. The rubella virus is as yet to the only virus that has been clearly associated with the development of diabetes and only congenital rubella infection is associated with diabetes. The 52 kd antigen appears to be expressed within beta cell granules 220.
37-38 kd
A number of groups have reported antibodies to 38 kd molecules in patients with Type 1A diabetes and their first-degree relatives. In the original description of antibodies precipitating the 64 kd molecule form islets, it was shown that some sera also precipitated a 38 kd molecule. Honeyman and coworkers 221 identified a 38 kd autoantigen as the nuclear transcription protein jun-B from an islet expression library. The relevance of this antigen to autoantibodies of prediabetics is unclear. An additional 38 kd molecule has been described by Pak and coworkers 222. Following their observation that some new-onset diabetic patients have cytomegalovirus infection, they immunized BALB/c mice with cytomegalovirus and obtained islet-reactive monoclonal antibodies that by Western blot recognize a 38 kd human islet protein.
Roep and coworkers 223 have identified a 38 kd islet granule autoantigen to which T cells react. The relationship between this antigen recognized by T cells and autoantibodies to 38 kd molecules is unknown.
155 kd
In 1990, McEvoy and coworkers used a novel method to detect diabetes-associated autoantigens. The developed a panel of mouse monoclonal antibodies reacting with the rat insulinoma cell line RIN5F and displaced the binding of the monoclonals to the tumor cells with diabetic sera. The binding of one of these monoclonals, termed 1A2, was selectively displaced by sera from diabetic children. The 1A2 monoclonal binds specifically to the insulinoma cell line RIN5F but not the other rat or human tissues. In a subsequent communication 223, the authors reported that monoclonal 1A2 recognizes by Western blot a 150 kd membrane-pound protein DAP1 (diabetes associated protein 1). Several sera from IDDM patients also identified a 150 kd band by Western blotting of rat brain homogenates. Using an assay based on displacement of the radiolabeled monoclonal binding to rat insulinoma cells, 159 of 169 sera of children with IDDM versus 10 of 351 age-matched control sera were positive for autoantibodies to DAP1. Autoantibodies to this antigen are, however, also present in a large percentage of nondiabetic first-degree relatives (> 60%).
GLIMA38
Baekkeskov and coworkers have immunoprecipitated a glycosylated islet cell membrane antigen which they have termed GLIMA 38(Glycated Islet cell Membrane-Associated protein). The molecule of 38 Kd was immunoprecipitated with sera from 19% of patients with new onset diabetes 224. Winnock and coworkers analyzed 100 new onset patients and 23 prediabetic siblings and found GLIMA 38 autoantibodies (by precipitation) in 38 and 35% respectively versus 0% in control subjects 225. In that almost all of the positive patients expressed another anti-islet autoantibody (e.g. IA-2 autoantibodies) they concluded that GLIMA 38 assay did not enhance the prediction or classification of diabetes.
Osteopontin and
Importin beta
Following the screening of a random peptide library antibodies were found that reacted with the molecule Osteopontin. Osteopontin is reported to be expressed in somatostatin producing islet cells. By radioassay patients with new onset diabetes did not differ from controls in the percentage of individuals with higher levels of osteopontin antibodies, with the ELISA assay found more positives than a radioassay 52. In a similar manner the molecule Importin beta was associated with autoimmunity with 60% of type 1 patients with ELISA assay and 30% of patients with other autoimmune disorders having antibodies 226. With a fluid phase radioassay, 6.3% of patients exceeded 99th percentile of normal controls with relatively low counts precipitated.
Nephrin, Densin and
Filtrin
A number of molecules are expressed in the kidney and in the islets including nephrin 227, densin and filtrin. Given the presence of the strong association of nephropathy with type 1A diabetes, sera from patients with type 1A diabetes were evaluated for autoantibodies to these molecules 55 using fluid phase radioimmunoprecipitation assay. Densin autoantibodies were detected in 33% of patients versus 2% of controls. Antibodies to filtrin were present in 11% versus 3% of controls.
CD38:ADP-ribosyl
cyclase/cADPR hydrolase
Several groups have described antibodies to CD38 in patients with type 2 diabetes and type 1 diabetes using either Western blot assays, or an enzymatic immunoassay on immobilized recombinant CD38 56, 228, 229. Prevalence of the reported autoantibodies are modest (e.g. 14% for type 2 patients versus 1% for controls) with lower positivity for type 1 diabetes.
SOX13(ICA12)
With an ELISA assay antibodies to SOX13, a transcription factor which is ICA12 50 was found in 18% of patients with type 1 diabetes, and a similar percentage of patients with hepatic autoimmunity 218, 230. The full length clone of SOX13 has been expressed and a series of patients evaluated for autoantibodies reacting with SOX13. 7.6% of new onset pediatric patients had anti-SOX13 antibodies exceeding the 99th percentile of normal controls, while no adult patient or adult control had such antibodies 49.
Glycolipid
Autoantigens
The initial studies suggesting that antibodies directed
against islet glycolipids may be a component of anti-islet autoantibodies came
from evaluation of monoclonal antibodies that reacted with neuronal
gangliosides and that also react with islets. Monoclonal antibodies A2B5 231, 3G5 232, R2D6 233, and tetanus toxin all react
with complex gangliosides and all give an ICA pattern of staining that involves
all cells within islets 130, 132.
In contrast to anti-islet monoclonal antibodies that react with proteins (e.g.,
HISL19 234, 235, the above
anti-ganglioside antibodies were not species specific 236. In that gangliosides are
composed of a ceramide group connected to a complex polysaccharide with at
least one sialic acid group, gangliosides are amphiphilic. Thus, organic solvents
such as methanol remove gangliosides from tissue sections, while gangliosides
are stable to nonpolar solvents such as acetone. Stability to acetone but
removal by methanol was a quality found for most
Glycolipids Islet Autoantigens include GT3 236, sulphatides 237, and what has been termed a GM2-1 238-240 ganglioside (on thin-layer chromatography it has a mobility between GM2 and GM1 standards). Despite the study of potential glycolipid islet autoantigens, no standard assay for diabetes associated autoantibodies reacting with these molecules has been developed.
Other Autoantigens
Reports from Elias and coworkers 41 suggested a role for T cell clones reacting to the heat shock protein (HSP) 65 in the pathogenesis of diabetes in the NOD mouse 241. In particular, these authors identified a peptide within the sequence of human HSP that was recognized by T cell clones that could transfer a transient form of diabetes and hyperglycemia 242. The administration of irradiation-attenuated HSP-specific T cells, or of the HSP peptide p277, prevented diabetes in the NOD mouse (controversial) or attenuated diabetes in the BB rat with a hydrolysed casein diet 243, 244. Despite initial reports, HSP65 is apparently not an autoantigen of humoral autoimmunity in man 245, though this is controversial 246. Studies of heat shock protein have progressed from animal studies 247 to phase I/phase II clinical trials with a web “report” of positive results in a trial of the peptide p277 in adult patients with new Phase III onset type 1 diabetes but apparently there was no effect in children 43.
In 1990, Johnson and coworkers 248, 249 suggested that an islet-specific glucose transporter is an autoantigen in Type 1A diabetes. The evidence supporting this conclusion derived from the ability of IgG fractions from diabetic patients to inhibit glucose uptake from rat islet cells. This inhibitory activity could be removed by incubation of the IgG fraction with islet cell and hepatocyte membranes (which express two types of glucose transporter, GLUT-1 and GLUT-2), but not with erythrocytes or brush border membranes (which express a only GLUT-2). Sera from 26 of 27 patients with IDDM inhibited the rat islet glucose uptake versus 0 of 5 sera from patients with NIDDM. Interestingly, the uptake of L-leucine was not affected by the IgG fractions. Though no direct biochemical data are provided for reaction of antibodies with GLUT-2, a publication indicates that introduction of the GLUT-2 gene into cell lines results in detection of surface autoantibodies 248, implying that GLUT-2 is recognized by autoantibodies of diabetic patients. Shehadeh and coworkers described autoantibodies reacting with CCL3 in patients with Type 1 diabetes 250. Regular proteins, which are associated with islet regeneration have been reported to be autoantigens251-253.
T Cell Reactivity
Many of the target molecules of autoantibodies described in this chapter have been reported to drive proliferation or cytokine secretion of T cells or their peptides can be used to produce tetramers reacting with T cells from animal models and patients with Type 1A diabetes or from islet autoantibody positive individuals4, 254-257.
In animal models a number of T cell assays, utilizing T cells obtained from spleen, pancreatic or peripheral lymph nodes, readily detect autoreactivity. For the autoantigen (IGRP) T cells are detectable in peripheral blood 258 and even can be imaged in vivo 259. Islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) was identified as the Beta-cell specific antigen targeted by a highly prevalent and pathogenic population of CD8+ T cells of NOD mice260-262. The IGRP peptide, amino acids 206-214, is naturally processed and presented and a tetramer with a high affinity peptide analogue was developed. A higher percentage of T lymphocytes in the circulation of NOD mice reacting with this NRP peptide (presented by K(d)) is associated with progression to diabetes 263, 264. A modified high affinity peptide termed NRP-V7 of the IGRP major CD8 autoantigen has been utilized to produce a class I tetramer. Despite this prominence knocking out the IGRP gene265 or inducing tolerance to IGRP 266, 267 does not alter progression to diabetes of NOD mice. Thus in some ways the immune response to IGRP may mark islet autoimmunity of the NOD similar to presence of insulin autoantibodies and not be a primary driver of disease. Nevertheless IGRP peptides can be utilized to drive CD8 T cells that can prevent diabetes 268, 269. Neither the NOD mouse nor man express anti-IGRP autoantibodies (Hutton et al, unpublished observations). Of note, autoreactivity to IGRP of NOD mice is “down-stream” of T cell reactivity to insulin269.
A putative autoantigen for T cells of the NOD mouse is dystrophia myotonica kinase, a widely distributed molecule that may be the target for AI4 cells 270. In that the molecule has been defined through studies primarily of mimotopes and it is widely distributed, one hypothesis is that the relevant islet autoantigen might not yet be defined, and DMK “is” a mimotope sequence.
Another well studied CD8 epitope is insulin peptide B:15-23 that appears to be an early epitope in insulitic lesions but not nearly as prominent as IGRP reactive T cells 271. Peakman and coworkers have described a CD8 T cell proinsulin epitope that is regulable by glucose 272. Roep evaluating anti-insulin CD8 T cell reaction cells with a tetramer assay 273 reported that high levels of such CD8 T cells predicted reoccurrence of diabetes of patients with new onset diabetes who received a bone marrow transplant. (FOCIS 2010, oral report).
Wegmann and coworkers found that a large percentage of CD4 T cells infiltrating islets of NOD mice strongly proliferate to insulin and, in particular, a B-chain peptide of insulin (B:9-23 274, 275 and the same peptide can be used to prevent diabetes) 276. In contrast, a minimal response of splenocytes to insulin is found. A subset of these T cell clones and their T cell receptors as retrogenesis are able to transfer diabetes into immunodeficient NOD mice160 and thus are sufficient for disease pathogenesis160. A response to insulin is seen as early as 4 weeks of age. The T cell clones of Wegmann and coworkers recognized insulin B-chain peptide B:9-23 and Unanue and coworkers have described that the B:9-23 peptide is likely produced in islets277. The majority of Wegmann T cell clones utilized a dominant Vα chain, Vα13.3 (also termed TRAV5D-4*04) 278 and T cells with this alpha chain as a Calpha only transgenic or retrogenic160 are able to induce insulin autoimmunity 199, 279. Alleva and coworkers have analyzed stimulation of human lymphocytes with the B:9-23 insulin peptide (identical sequence in mouse and man insulin) and find reactivity amongst patients but not controls 280. Kappler and coworkers discovered that the insulin peptide B:9-23 is bound in a specific register of the I-Ag7 class II molecule including the T cell clones reported by Uananue and coworkers281. This is a low affinity register (termed register 3) with arginine of the peptide binding to pocket 9 (if the arginine of the peptide is present and the class II groove filled). This discovery combined with fixing the peptide in register3 has led to the development of tetramers to detect T cells reacting with the insulin B:9-23 peptide159. Approximately 4% of T cells invading islets of NOD mice react with these register fixed tetramers159. In addition von Boehmer and coworkers have used this knowledge to use a B:9-23 mimetope with glutamic acid substituting for the arginine binding in pocket 9 to potently prevent diabetes of NOD mice282.
Kent and coworkers isolated T cell clones reacting with an insulin A chain determinant from pancreatic lymph nodes of man 283. In the NOD mouse model with an insulin peptide tetramer (B chain amino acids 15 to 23) autoreactive T cells can be detected particularly within islets 284. In addition to insulin peptides, proinsulin peptides and in particular a peptide spanning the B chain C-peptide junction is a target of autoimmunity 285-288. Durinovic-Belló utilizing tetramer assay has reported that levels of high avidity proinsulin peptide reactive T cells are decreased in individuals with the low risk insulin gene VNTR 289.
In addition tetramers have been developed utilizing the mimotopes of the BDC2.5 clone that responds to a chromagranin peptide 290. This clone and transgenic have provided a wealth of data concerning the pathogenesis of diabetes of the NOD mouse 291-293. The target of BDC 2.5 has been identified as chromagranin, with evidence for a specific peptide that does not completely fill the presenting I-Ag7 groove294, 295.
Subtractive hybridization of islets of a patient with new onset diabetes led to the identification of a CDNA encoding hepatocarcinoma - intestine-pancreas/pancreatic associated protein (HIP/PAP) for which a T cell response from NOD mice was identified 296. Reg II was predominantly expressed in islets and vaccination with C or N terminal could accelerate or inhibit NOD diabetes 36.
Atkinson and coworkers 297, 298 studied the reactivity of peripheral blood mononuclear cells to recombinant glutamic acid decarboxylase in newly diagnosed type 1 patients, ICA-positive and ICA-negative relatives of individuals with diabetes, and in healthy controls. A higher proportion of subjects gave a positive proliferative response to GAD among the newly diagnosed patients than among the controls. Interestingly, nondiabetic ICA-positive relatives were also more likely to express T cell reactivity to GAD (63% versus 11%). Harrison and coworkers 299 reported proliferation of peripheral blood T cells in presence of the central region of GAD67 (aa 208-404) in 38% of newly diagnosed IDDM patients and in 41% of ICA-positive relatives (> 20 JDF units). Only 4% of HLA-matched controls responded to GAD67. In a subsequent report, Harrison and coworkers described an assay utilizing fluorescent DNA labeling (fluorescent dye 5,6-carboxylfluorescein diacetate succinimidyl ester[CFSE) 300.
Assays for reactivity of peripheral blood lymphocytes with GAD give relatively low stimulation indices 301. In the NOD mouse, utilizing spleen cells, Tisch and coworkers 302 reported that T cell proliferative responses to GAD precede responses to other antigens. The stimulation indices again are low but of interest and T cell clones to GAD have been derived 303. Nepom and coworkers have isolated a peptide of GAD65 and have developed T cell tetramer (DRB1*0401) assays 304 for reactivity with this epitope 305, and tetramer assays have evaluated responses post pancreatic transplantation 306.
The study to date of T cell responses of man to islet autoantigens has been particularly difficult with relatively low levels of stimulation, when stimulation is observed. This may relate to a very low frequency of autoreactive T cells outside of the islets. In an international workshop with multiple laboratories studying a series of "blinded" islet antigens and peptides none of the laboratories could distinguish a small panel of patients with diabetes from control individuals 307, 308. We believe it is likely that the assay formats will have to be greatly improved to be able to reliably detect T cell responses and there has been recent progress in particular for CD8 T lymphocytes 272, 309, 310 and with register fixed class II tetramers159
Testing proliferation to multiple autoantigens by Dosch and coworkers and proliferation to multiple bands on polyacrylamide gel chromatography of human islets by Brooks-Worrell and coworkers indicated that combining responses significantly distinguished control and patient samples 311. The general approach of adding responses to multiple different antigens/antigenic determinants allows distinction between control and patient samples, though with specificities usually are far below that of most autoantibody assays 311.
Characterization of T cell reactivity has been particularly difficult in man and we believe this results from lack of ability to obtain T lymphocytes from the site of pathology, namely pancreatic islets. In an effort to obtain access to such T cells, we piloted a program where cadaveric organ donors are screened in real time for the expression of anti-islet autoantibodies 312. Between 1/300 and 1/400 of such donors will express multiple anti-islet autoantibodies 312, 313 and we predict that the pancreas from such individuals (that can be obtained at the time of organ donation), will harbor relevant T cell clones. The nPOD program (www.jdrfnpod.org ) now has an international program to obtain the pancreas of patients with Type I and “pre-Type I” diabetes and provide tissue and web access to histology 314.
Figure 10.11:
Development of anti-islet autoantibodies following islet transplantation.
Islet Transplantation
With the initial success of the "Edmonton
Protocol" transplantation of islets has increased, especially in patients
with severe recurrent hypoglycemia on insulin therapy for type 1A diabetes 315. With follow up most patients unfortunately
eventually require resumption of insulin therapy. For both islet and pancreatic
transplantation there is evidence that recurrent anti-islet autoimmunity may
contribute to failure of grafts to reverse hyperglycemia or for patients with
stable grafts to lose function and in particular lose insulin independence. As
illustrated in Figure 10.11 a subset of patients (approximately 10%) with islet
transplantation can have a dramatic rise in islet autoantibodies and this has
been associated with the loss of the graft 315. Studies of the
influence of anti-islet autoantibodies (with determination of biochemical
anti-islet autoantibodies) on graft function were sponsored by the Immune
Tolerance Network as part of the evaluation of the
Conclusions
Investigators have defined a growing family of islet
autoantigens recognized by autoantibodies of prediabetics. It is likely that
such a large number of autoantibodies are secondary to beta cell destruction
and the presentation of these antigens in an inflammatory environment.
Nevertheless, studies in the NOD mouse suggest that influencing the immune
response to several autoantigens (e.g., oral insulin, intravenous GAD,
subcutaneous insulin) can prevent diabetes. At present, the primacy in disease
pathogenesis of any of the defined autoantigens is unclear in man, but with considerable
evidence that insulin may a primary autoantigen of the NOD mouse and perhaps
man 161, 267.
Figure 10.12: GAD65
and IA-2(ICA512bdc) autoantibodies of the DPT-1 (Diabetes Prevention Trial-1
study.
Despite uncertainty about pathogenic importance, the ability to biochemically detect autoantibodies to a series of autoantigens greatly facilitate the prediction of Type 1A diabetes (Figures 10.12 and10.13). Utilizing only four radioassays for autoantibodies (insulin, GAD, IA-2 and ZnT8) 95% of prediabetic or recent-onset diabetic children express one or more antibodies (versus only 4 out of 100 controls). More than 80% express two or more autoantibodies, versus "no” controls. Approximately 25% of diabetics or prediabetics express all of the above autoantibodies. The expression of autoantibodies assorts independently in prediabetic individuals and thus, by utilizing the four assays, a high positive predictive value for disease is possible while retaining disease sensitivity (Combinatorial Autoantibody Prediction) 27, 318, 319.
Figure 10.13:
Combinatorial Autoantibody prediction (Modified from Verge et al. Diabetes
47:1857-1866, 1998)
"Biochemical" autoantibody screening will likely have its largest impact for the prediction of Type 1A diabetes in the general population where the positive predictive value of ICA positivity is lower than201 5%, while presence of two or more of four major autoantibodies in children followed from birth for more than a decade gives positive predictive values greater than 90%(DAISY unpublished). A recent study of the general population from Finland with up to 27 years of follow-up indicates that the presence of both GAD and IA-2 autoantibodies together identify 60% of prediabetics with a positive predictive value of 100% 320. Screening of the general population for diabetes risk is likely to be a major importance if trials for the prevention of Type 1A diabetes among first-degree relatives are successful. Approximately 90% of individuals who develop Type 1A diabetes have no first-degree relatives with the disease and thus such screening may be essential for the large-scale prevention of Type 1A diabetes.
(Figure 10.14) illustrates a number of caveats of screening
for anti-islet autoantibodies in "low" risk populations. (In this case, relatives of patients with
type 1 diabetes and HLA characterized individuals from the general population
with HLA determined increased risk [e.g. DR3/DR4-DQ8] of diabetes [more than
30,000 newborns from the general population were HLA typed from cord blood in
the DAISY study to identify those with high risk genotypes]) 66.
Overall both groups (HLA identified general population and first degree
relatives without HLA typing) have an
approximate risk of diabetes of 1/20 compared to a
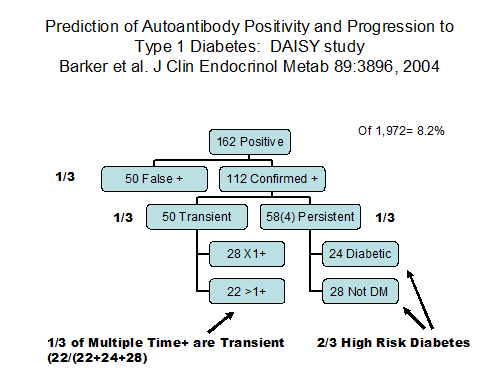
Figure 10.14:
Confirmation and Persistence of islet autoantibodies (GAD65,IA-2,insulin).
We are at a somewhat unusual phase in the application of our knowledge of humoral autoimmunity of type 1A diabetes. Namely we can predict the disease, identifying varying degrees of diabetes risk, such that trials for the prevention of type 1A diabetes are feasible. Unfortunately at present we do not have a proven and safe therapy to delay or prevent type 1A diabetes. As analyzed in a number of studies increased anxiety is associated with screening for serious disorders 321, 322, but most of the studies that have addressed this issue have found relatively mild and transient increases in such anxiety with islet autoantibody screening 323-325. Balancing such an increase in anxiety at present, it is very clear that screening for anti-islet autoantibodies coupled with metabolic and close monitoring dramatically decreases the development of ketoacidosis at diabetes diagnosis, such that only one child of thirty in the DAISY study required hospitalization versus 40% of children in the general population presenting with diabetes 326. This dramatic difference is almost certainly due to earlier diagnosis and many of the children from the general population had glucoses greater than 1,000 mg% while none of the DAISY children had glucose greater than 400mg% at diagnosis. Though there is clinical benefit in earlier diagnosis, the major impetus for identifying at risk individuals will be driven by trials for disease prevention. It is likely that islet autoantibodies will always be a useful, though incomplete reflection of pathogenesis, and assays for autoreactive pathogenic T lymphocytes are essential to accelerate the pace of clinical trials. International networks are now available to foster translational research in the area of type 1A diabetes, with Trialnet and the Immune Tolerance Network, both of which have web pages and an open solicitation for development of novel trials and novel assays.
Reference
List
1. Bottazzo GF, Florin-Christensen A, Doniach D.
Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine
deficiencies. Lancet 1974;2(7892):1279-1283.
2. Palmer JP, Asplin CM, Clemons P et al.
Insulin antibodies in insulin-dependent diabetics before insulin treatment.
Science 1983;222(4630):1337-1339.
3. DiLorenzo TP. Multiple antigens versus single
major antigen in type 1 diabetes: arguing for multiple antigens. Diabetes Metab
Res Rev 2011;27(8):778-783.
4. Roep BO, Peakman M. Antigen targets of type 1
diabetes autoimmunity. Cold Spring Harb Perspect Med 2012;2(4):a007781.
5. Lampasona V, Schlosser M, Mueller PW et al.
Diabetes antibody standardization program: first proficiency evaluation of
assays for autoantibodies to zinc transporter 8. Clin Chem
2011;57(12):1693-1702.
6. Parikka V, Nanto-Salonen K, Saarinen M et al.
Early seroconversion and rapidly increasing autoantibody concentrations predict
prepubertal manifestation of type 1 diabetes in children at genetic risk.
diabetol 2012.
7. Vehik K, Beam CA, Mahon JL et al. Development
of autoantibodies in the TrialNet Natural History Study. Diab care
2011;34(9):1897-1901.
8. Miao D, Yu L, Eisenbarth GS. Role of
autoantibodies in type 1 diabetes. Front Biosci 2007;12:1889-98.:1889-1898.
9. Isermann B, Ritzel R, Zorn M, Schilling T,
Nawroth PP. Autoantibodies in diabetes mellitus: current utility and
perspectives. Exp Clin Endocrinol Diabetes 2007;115(8):483-490.
10. Greenbaum C, Palmer JP, Kuglin B, Kolb H, and
Participating Laboratories. Insulin autoantibodies measured by radioimmunoassay
methodology are more related to insulin-dependent diabetes mellitus than those
measured by enzyme-linked immunosorbent assay: results of the Fourth
International Workshop on the Standardization of Insulin Autoantibody
Measurement. J Clin Endocrinol Metab 1992;74(5):1040-1044.
11. Baekkeskov S, Aanstoot H-J, Christgau S et
al. Identification of the 64K autoantigen in insulin-dependent diabetes as the
GABA-synthesizing enzyme glutamic acid decarboxylase [published erratum appears
in Nature 1990 Oct 25;347(6295):782]. Nature 1990;347(6289):151-156.
12. Rabin DU, Pleasic SM, Palmer-Crocker R,
Shapiro JA. Cloning and expression of IDDM-specific human autoantigens. diab
1992;41(2):183-186.
13. Rabin DU, Pleasic SM, Shapiro JA et al. Islet
cell antigen 512 is a diabetes-specific islet autoantigen related to protein
tyrosine phosphatases. J Immunol 1994;152(6):3183-3188.
14. Lan MS, LU J, Goto Y, Notkins AL. Molecular
cloning and identification of a receptor-type protein tyrosine phosphatase,
IA-2, from human insulinoma. DNA Cell Biol 1994;13(5):505-514.
15. Wasmeier C, Hutton JC. Molecular cloning of
phogrin, a protein-tyrosine phosphatase homologue localized to insulin
secretory granule membranes. J Biol Chem 1996;271(30):18161-18170.
16. LU J, Li Q, Xie H et al. Identification of a
second transmembrane protein tyrosine phosphatase, IA-2b, as an autoantigen in insulin-dependent diabetes mellitus: precursor of
the 37-kDa tryptic fragment. Proc Natl Acad Sci USA 1996;93:2307-2311.
17. Wenzlau JM, Moua O, Sarkar SA et al. SlC30A8
is a major target of humoral autoimmunity in type 1 diabetes and a predictive
marker in prediabetes. Ann N Y Acad Sci 2008;1150:256-259.
18. Wenzlau JM, Juhl K, Yu L et al. The cation
efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1
diabetes. Proc Natl Acad Sci U S A 2007;104(43):17040-17045.
19. Burbelo PD, Hirai H, Issa AT et al.
Comparison of radioimmunoprecipitation with luciferase immunoprecipitation for
autoantibodies to GAD65 and IA-2{beta}. Diab care 2010.
20. Yu L, Miao D, Scrimgeour L, Johnson K, Rewers
M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with
differential risk: nonradioactive bivalent proinsulin/insulin autoantibody
assay. diab 2012;61(1):179-186.
21. Schlosser M, Mueller PW, Torn C, Bonifacio E,
Bingley PJ. Diabetes Antibody Standardization Program: evaluation of assays for
insulin autoantibodies. diabetol 2010;53(12):2611-2620.
22. Achenbach P, Koczwara K, Knopff A, Naserke H,
Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin
anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest
2004;114(4):589-597.
23. Steck AK, Johnson K, Barriga KJ et al. Age of
islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2
autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes
autoimmunity study in the young. Diab care 2011;34(6):1397-1399.
24. Curnock RM, Reed CR, Rokni S, Broadhurst JW,
Bingley PJ, Williams AJ. 'Insulin autoantibody affinity measurement using a
single concentration of unlabelled insulin competitor discriminates risk in
relatives of patients with type 1 diabetes. Clin Exp Immunol 2012;167(1):67-72.
25. Yu L, Boulware DC, Beam CA et al. Zinc
Transporter-8 Autoantibodies Improve Prediction of Type 1 Diabetes in Relatives
Positive for the Standard Biochemical Autoantibodies. Diab care 2012.
26. Bingley PJ, Williams AJ, Colman PG et al.
Measurement of islet cell antibodies in the Type 1 Diabetes Genetics
Consortium: efforts to harmonize procedures among the laboratories. Clin Trials
2010;7(1 Suppl):S56-S64.
27. Verge CF, Gianani R, Kawasaki E et al.
Prediction of type I diabetes in first-degree relatives using a combination of
insulin, GAD, and ICA512bdc/IA-2 autoantibodies. diab 1996;45(7):926-933.
28. Bingley PJ. Clinical applications of diabetes
antibody testing. J Clin Endocrinol Metab 2010;95(1):25-33.
29. Orban T, Sosenko JM, Cuthbertson D et al.
Pancreatic Islet Autoantibodies as Predictors of Type 1 Diabetes in the
Diabetes Prevention Trial-Type 1 (DPT-1). Diab care 2009.
30. Kemp EH, Emhemad S, Akhtar S, Watson PF,
Gawkrodger DJ, Weetman AP. Autoantibodies against tyrosine hydroxylase in
patients with non-segmental (generalised) vitiligo. Exp Dermatol
2011;20(1):35-40.
31. Thomas NM, Ginsberg-Fellner F, Mcevoy RC.
Strong association between diabetes and displacement of mouse anti-rat
insulinoma cell monoclonal antibody by human serum in vitro. diab
1990;39:1203-1211.
32. Pan
Y-X, Thomas NM, Mcevoy RC. Purification and partial characterization of 160 kD
and 155 kD B cell membrane antigens detected by MAb 1A2. Autoimmunity
15,Supplement, 77A. 1993.
Ref Type: Abstract
33. Karounos DG, Wolinsky JS, Thomas JW.
Monoclonal antibody to rubella virus capsid protein recognizes a b-cell antigen. J Immunol 1993;150:3080-3085.
34. Arden SD, Zahn T, Steegers S et al. Molecular
cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic
subunit-related protein. diab 1999;48(3):531-542.
35. Canino GJ, Bird HR, Shrout PE et al. The
prevalence of specific psychiatric disorders in Puerto Rico. Arch Gen
Psychiatry 1987;44(8):727-735.
36. Gurr W, Shaw M, Li Y, Sherwin R. RegII is a
beta-cell protein and autoantigen in diabetes of NOD mice. Diabetes
2007;56(1):34-40.
37. Dosch HM, Karjalainen J, VanderMeulen J. Lack
of immunity to bovine serum albumin in insulin-dependent diabetes mellitus
(letter). N Engl J Med 1994;330(22):1616-1617.
38. Pietropaolo M, Castano L, Babu S et al. Islet
cell autoantigen 69 kDa (ICA69): molecular cloning and characterization of a
novel diabetes associated autoantigen. J Clin Invest 1993;92:359-371.
39. Gordon TP, Cavill D, Neufing P, Zhang YJ,
Pietropaolo M. ICA69 autoantibodies in primary Sjogren's syndrome. Lupus
2004;13(6):483-484.
40. Spitzenberger F, Pietropaolo S, Verkade P et
al. Islet cell autoantigen of 69 kDa is an arfaptin-related protein associated
with the Golgi complex of insulinoma INS-1 cells. J Biol Chem
2003;278(28):26166-26173.
41. Elias D, COHEN I, SHECHTER Y, Spirer Z, Golander
A. Antibodies to insulin receptor followed by anti-idiotype. diab
1987;36:348-354.
42. Birk OS, Elias D, Weiss AS et al. NOD mouse
diabetes: the ubiquitous mouse hsp60 is a beta-cell target antigen of
autoimmune T cells. J Autoimmun 1996;9(2):159-166.
43. Huurman VA, van der Meide PE, Duinkerken G et
al. Immunological efficacy of heat shock protein 60 peptide DiaPep277 therapy
in clinical type I diabetes. Clin Exp Immunol 2008;152(3):488-497.
44. Atkinson MA, Maclaren NK, Scharp DW. No
evidence for HSP65 in insulin dependent diabetes. Lancet 1990;337:263-264.
45. Chang YH, Shiau MY, Tsai ST, Lan MS.
Autoantibodies against IA-2, GAD, and topoisomerase II in type 1 diabetic
patients. Biochem Biophys Res Commun 2004;320(3):802-809.
46. Colman PG, Nayak RC, Campbell IL, Eisenbarth
GS. Binding of cytoplasmic islet cell antibodies is blocked by human pancreatic
glycolipid extracts. diab 1988;37(5):645-652.
47. Pfleger C, Kaas A, Hansen L et al. Relation
of circulating concentrations of chemokine receptor CCR5 ligands to C-peptide,
proinsulin and HbA1c and disease progression in type 1 diabetes. Clin Immunol
2008;128(1):57-65.
48. Aanstoot H-J, Kang S-M, Kim J et al.
Identification and characterization of glima 38, a glycosylated islet cell membrane
antigen, which together with GAD65 and IA2 marks the early phases of autoimmune
response in type I diabetes. J Clin Invest 1996;97(12):2772-2783.
49. Lampasona V, Scirpoli M, Bosi E, Bonifacio E.
ICA12(SOX13) autoantibodies are unlikely to be a useful marker for pre-clinical
Type I diabetes. diabetol 2001;44(2):267.
50. Gupta M, Tandon N, Shtauvere-Brameus A,
Sanjeevi CB. ICA12 autoantibodies are associated with non-DR3/non-DR4 in
patients with latent autoimmune diabetes in adults from northern India. Ann N Y
Acad Sci 2002;958:329-332.
51. Lernmark Å, Freedman ZR, Hofmann C et al.
Islet-cell-surface antibodies in juvenile diabetes mellitus. N Engl J Med
1978;299:375-380.
52. Fierabracci A, Biro PA, Yiangou Y et al.
Osteopontin is an autoantigen of the somatostatin cells in human islets:
identification by screening random peptide libraries with sera of patients with
insulin-dependent diabetes mellitus. Vaccine 1999;18(3-4):342-354.
53. Ola TO, Biro PA, Hawa MI et al. Importin
beta: A novel autoantigen in human autoimmunity identified by screening random
peptide libraries on phage. J Autoimmun 2006;26(3):197-207.
54. Winer S, Tsui H, Lau A et al. Autoimmune
islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive.
Nat Med 2003;9(2):198-205.
55. Rinta-Valkama J, Aaltonen P, Lassila M et al.
Densin and filtrin in the pancreas and in the kidney, targets for humoral
autoimmunity in patients with type 1 diabetes. Diabetes Metab Res Rev 2006;.
56. Mallone R, Perin PC. Anti-CD38 autoantibodies
in type? diabetes. Diabetes Metab Res Rev 2006;.
57. Hu CY, Rodriguez-Pinto D, Du W et al.
Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes
in mice. J Clin Invest 2007;117(12):3857-3867.
58. Greeley SA, Katsumata M, Yu L et al.
Elimination of maternally transmitted autoantibodies prevents diabetes in
nonobese diabetic mice. Nat Med 2002;8(4):399-402.
59. Ziegler A-G, Hummel M, Schenker M, Bonifacio
E. Autoantibody appearance and risk for development of childhood diabetes in
offspring of parents with type 1 diabetes. The 2-year analysis of the German
BABYDIAB study. diab 1999;48:460-468.
60. Roll U, Christie MR, Füchtenbusch M, Payton
MA, Hawkes CJ, Ziegler AG. Perinatal autoimmunity in offspring of diabetic
parents. The German Multi-Center BABY-DIAB study: detection of humoral immune
responses to islet antigens in early childhood. diab 1996;45(7):967-973.
61. Martin S, Wolf-Eichbaum D, Duinkerken G et
al. Development of type 1 diabetes despite severe hereditary B-lymphocyte
deficiency. N Engl J Med 2001;345(14):1036-1040.
62. Mcevoy RC, Thomas NM, Ness J. Humoral immune
markers: additional islet cell antigens--important clues to pathogenesis or red
herrings? In: Palmer JP, editor. Prediction, prevention, and genetic counseling
in IDDM. Chichester, England: Wiley; 1996:97-108.
63. DiLorenzo TP, Graser RT, Ono T et al. Major
histocompatibility complex class I-restricted T cells are required for all but
the end stages of diabetes development in nonobese diabetic mice and use a
prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci
USA 1998;95(21):12538-12543.
64. YALOW RS, Berson SA. Assay of plasma insulin
in human subjects by immunological methods. Nature 1959;184 (Suppl
21):1648-1649.
65. Babaya N, Liu E, Miao D, Li M, Yu L,
Eisenbarth GS. Murine high specificity/sensitivity competitive europium insulin
autoantibody assay. Diabetes Technol Ther 2009;11(4):227-233.
66. Barker JM, Barriga K, Yu L et al. Prediction
of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young
(DAISY). J Clin Endocrinol Metab 2004;89:3896-3902.
67. Falorni A, Örtqvist E, Persson B, Lernmark Å.
Radioimmunoassays for glutamic acid decarboxylase (GAD65) and GAD65
autoantibodies using 35S or 3H recombinant human ligands.
J Immunol Methods 1995;186:89-99.
68. Kawasaki E, Eisenbarth G. High-Throughput
Radioassays for Autoantibodies to Recombinant Autoantigens. Frontiers in
Bioscience 2000;5:181-190.
69. Liu E, Eisenbarth GS. Accepting clocks that
tell time poorly: fluid-phase versus standard ELISA autoantibody assays. Clin
Immunol 2007;125(2):120-126.
70. Yu L, Wang J, O'Dell JR, Oates J, Arend WP,
Eisenbarth GS. Anti-dsDNA antibody assay: high specificity and sensitivity with
a filtration radioassay in comparison to low specificity with the standard
ELISA. J Rheumatol 2007;34(4):734-739.
71. Bao F, Yu L, Babu S et al. One third of HLA
DQ2 homozygous patients with type 1 diabetes express celiac disease associated
transglutaminase autoantibodies. J Autoimmunity 1999;13:143-148.
72. Liu E, Li M, Bao F et al. Need for
quantitative assessment of transglutaminase autoantibodies for celiac disease
in screening-identified children. J Pediatr 2005;146(4):494-499.
73. Falorni A, Nikoshkov A, Laureti S et al. High
diagnostic accuracy for idiopathic Addison's disease with a sensitive
radiobinding assay for autoantibodies against recombinant human 21-hydroxylase.
J Clin Endocrinol Metab 1995;80(9):2752-2755.
74. Laureti S, Aubourg P, Calcinaro F et al.
Etiological diagnosis of primary adrenal insufficiency using an original
flowchart of immune and biochemical markers. J Clin Endocrinol Metab
1998;89(9):3163-3168.
75. Yu L, Brewer KW, Gates S et al. DRB1*04 and
DQ alleles: expression of 21-hydroxylase autoantibodies and risk of progression
to Addison's disease. J Clin Endocrinol Metab 1999;84(1):328-335.
76. Bonifacio E, Yu L, Williams AK et al.
Harmonization of glutamic acid decarboxylase and islet antigen-2 autoantibody
assays for national institute of diabetes and digestive and kidney diseases
consortia. J Clin Endocrinol Metab 2010;95(7):3360-3367.
77. Rulli M, Kuusisto A, Salo J, Kojola H, Simell
O. Time-resolved fluorescence imaging in islet cell autoantibody quantitation.
J Immunol Methods 1997;208(2):169-179.
78. Hagopian WA, Sanjeevi CB, Kockum I et al.
Glutamate decarboxylase-, insulin-, and islet cell-antibodies and HLA typing to
detect diabetes in a general population-based study of Swedish children. J Clin
Invest 1995;95:1505-1511.
79. Brooking H, Ananieva-Jordanova R, Arnold C et
al. A sensitive non-isotopic assay for GAD65 autoantibodies. Clin Chim Acta
2003;331(1-2):55-59.
80. Valdez SN, Iacono RF, Villalba A, Cardoso LA,
Ermacora MR, Poskus E. A radioligand-binding assay for detecting antibodies
specific for proinsulin and insulin using 35S-proinsulin. J Immunol Methods
2003;279(1-2):173-181.
81. Lo B, Swafford AD, Shafer-Weaver KA et al.
Antibodies against insulin measured by electrochemiluminescence predicts
insulitis severity and disease onset in non-obese diabetic mice and can
distinguish human type 1 diabetes status. J Transl Med 2011;9:203.
82. Nerup J. Addison's disease - clinical
studies: A report of 108 cases. Acta
Endocrinol 1974;76:127-141.
83. Eizirik DL, Sandler S, Palmer JP. Repair of
pancreatic b-cells: a relevant phenomenon in early IDDM? diab
1993;42:1383-1391.
84. Thai A-C, Eisenbarth GS. Natural history of
IDDM. Diabetes Rev 1993;1:1-14.
85. Schatz DA, Atkinson MA. Islet cell
autoantibodies: a case of a premature obituary. Pediatr Diabetes
2005;6(4):181-183.
86. Pietropaolo M, Yu S, Libman IM et al.
Cytoplasmic islet cell antibodies remain valuable in defining risk of
progression to type 1 diabetes in subjects with other islet autoantibodies.
Pediatr Diabetes 2005;6(4):184-192.
87. Turner R, Stratton I, Horton V et al. UKPDS
25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction
of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group.
Lancet 1997;350(9087):1288-1293.
88. Bottazzo GF, Bosi E, Cull CA et al. IA-2
antibody prevalence and risk assessment of early insulin requirement in
subjects presenting with type 2 diabetes (UKPDS 71). diabetol
2005;48(4):703-708.
89. Desai M, Cull CA, Horton VA et al. GAD
autoantibodies and epitope reactivities persist after diagnosis in latent
autoimmune diabetes in adults but do not predict disease progression: UKPDS 77.
Diabetologia 2007;.
90. Maioli M, Pes GM, Delitala G et al. Number of
autoantibodies and HLA Genotype, more than high titers of glutamic acid
decarboxylase autoantibodies (GADA65Ab), predict insulin dependence in latent
autoimmune diabetes of adults (LADA). Eur J Endocrinol 2010.
91. Zimmet P. Antibodies to glutamic acid
decarboxylase in the prediction of insulin dependency. [Review] [31 refs].
Diabetes Res Clin Pract 1996;34 Suppl:S125-31.
92. Lampasona V, Petrone A, Tiberti C et al. Zinc
transporter 8 antibodies complement GAD and IA-2 antibodies in the
identification and characterization of adult-onset autoimmune diabetes: Non
Insulin Requiring Autoimmune Diabetes (NIRAD) 4. Diab care 2010;33(1):104-108.
93. Desai M, Clark A. Autoimmune diabetes in
adults: lessons from the UKPDS. Diabet Med 2008;25 Suppl 2:30-34.
94. Klingensmith GJ, Pyle L, Arslanian S et al.
The Presence of GAD and IA-2 Antibodies in Youth with a Type 2 Diabetes
Phenotype: Results from the TODAY Study. Diab care 2010.
95. Greenbaum CJ, Palmer JP, Nagataki S et al.
Improved specificity of ICA assays in the Fourth International Immunology of
Diabetes Exhange Workshop. diab 1992;41:1570-1574.
96. Bingley PJ, Bonifacio E, Mueller PW. Diabetes
antibody standardization program: first assay proficiency evaluation. diab
2003;52(5):1128-1136.
97. Achenbach P, Schlosser M, Williams AJ et al.
Combined testing of antibody titer and affinity improves insulin autoantibody
measurement: Diabetes Antibody Standardization Program. Clin Immunol
2007;122(1):85-90.
98. Henderson
LO, Atkinson M, Bingley PJ et al. Dried blood-spots as a matrix for Type-1
diabetes autoantibody detection. Diabetes 49 (Suppl 1)[A36]. 2000.
Ref Type: Abstract
99. Krischer JP, Cuthbertson DD, Yu L et al.
Screening strategies for the identification of multiple antibody-positive
relatives of individuals with type 1 diabetes. J Clin Endocrinol Metab
2003;88(1):103-108.
100. Gianani R, Pugliese A, Bonner-Weir S et al.
Prognostically significant heterogeneity of cytoplasmic islet cell antibodies
in relatives of patients with type I diabetes. diab 1992;41(3):347-353.
101. Genovese S, Bonifacio E, McNally JM et al.
Distinct cytoplasmic islet cell antibodies with different risks for type I
(insulin-dependent) diabetes mellitus. diabetol 1992;35:385-388.
102. Sosenko JM, Mahon J, Rafkin L et al. A
comparison of the baseline metabolic profiles between Diabetes Prevention
Trial-Type 1 and TrialNet Natural History Study participants. Pediatr Diabetes
2010.
103. Solimena M, Folli F, Denis-Donini S et al.
Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man
syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med
1988;318(16):1012-1020.
104. Verge CF, Stenger D, Bonifacio E et al. Combined
use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin
autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes:
Combinatorial Islet Autoantibody Workshop. diab 1998;47(12):1857-1866.
105. Bingley PJ, Christie MR, Bonifacio E et al.
Combined analysis of autoantibodies improves prediction of IDDM in islet cell
antibody-positive relatives. diab 1994;43(11):1304-1310.
106. Achenbach P, Bonifacio E, Williams AJ,
Ziegler AG, Gale EA, Bingley PJ. Autoantibodies to IA-2beta improve diabetes
risk assessment in high-risk relatives. Diabetologia 2008;.
107. Achenbach P, Warncke K, Reiter J et al.
Stratification of type 1 diabetes risk on the basis of islet autoantibody
characteristics. diab 2004;53(2):384-392.
108. Park YS, Kawasaki E, Kelemme K et al. Humoral
autoreactivity to an alternative spliced variant of ICA512/IA2 in type 1
diabetes. diabetol 2000;50:895-900.
109. Torn C, Mueller PW, Schlosser M, Bonifacio E,
Bingley PJ. Diabetes Antibody Standardization Program: evaluation of assays for
autoantibodies to glutamic acid decarboxylase and islet antigen-2. diabetol
2008;51(5):846-852.
110. Mueller PW, Bingley PJ, Bonifacio E,
Steinberg KK, Sampson EJ. Predicting type 1 diabetes using autoantibodies: the
latest results from the diabetes autoantibody standardization program. Diabetes
Technol Ther 2002;4(3):397-400.
111. Erdo SL, Wolff JR. Gamma-aminobutyric acid
outside the mammalian brain. J Neurochem 1990;54:363-372.
112. Kanaani J, Diacovo MJ, El Husseini A, Bredt
DS, Baekkeskov S. Palmitoylation controls trafficking of GAD65 from Golgi
membranes to axon-specific endosomes and a Rab5a-dependent pathway to
presynaptic clusters. J Cell Sci 2004;117(Pt 10):2001-2013.
113. Petersen JS, Russel S, Marshall MO et al.
Differential expression of glutamic acid decarboxylase in rat and human islets.
diab 1993;42:484-495.
114. Kim J, Richter W, Aanstoot HJ et al.
Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic
islets. diab 1993;42(12):1799-1808.
115. Eisenbarth GS. Mouse or man: is GAD the cause
of type I diabetes? Diab care 1994;17:1-3.
116. Burton AR, Baquet Z, Eisenbarth GS et al.
Central Nervous System Destruction Mediated by Glutamic Acid
Decarboxylase-Specific CD4+ T Cells. J Immunol 2010.
117. Christie M, Landin-Olsson M, Sundkvist G,
Dahlquist G, Lernmark Å, Baekkeskov S. Antibodies to a Mr-64,000 islet cell
protein in Swedish children with newly diagnosed type I (insulin-dependent)
diabetes. diabetol 1988;31(8):597-602.
118. Baekkeskov S, Landin M, Kristensen JK et al.
Antibodies to a 64,000 Mr human islet cell antigen precede the clinical onset
of insulin-dependent diabetes. J Clin Invest 1987;79:926-934.
119. Hayakawa N, Premawardhana LD, Powell M et al.
Isolation and characterization of human monoclonal autoantibodies to glutamic
acid decarboxylase. Autoimmunity 2002;35(5):343-355.
120. Gianani
R, Jackson R, Eisenbarth GS. Evidence that the autoantigen of restricted ICA is
GAD. Diabetes Res Clin Prac 14 (suppl. 1), S13. 1991.
Ref Type: Abstract
121. Velloso LA, Kampe O, Hallberg A, Christmanson
L, Betsholtz C, Karlsson FA. Demonstration of GAD-65 as the main immunogenic
isoform of glutamate decarboxylase in type 1 diabetes and determination of
autoantibodies using a radioligand produced by eukaryotic expression. J Clin
Invest 1993;91:2084-2090.
122. Marcus P, Yan X, Bartley B, Hagopian W. LIPS
islet autoantibody assays in high-throughput format for DASP 2010. Diabetes
Metab Res Rev 2011;27(8):891-894.
123. Harrison LC, Honeyman MC, DeAizpurua HJ et
al. Inverse relation between humoral and cellular immunity to glutamic acid
decarboxylase in subjects at risk of insulin-dependent diabetes. Lancet
1993;341(8857):1365-1369.
124. Bingley PJftIG. Interactions of age, islet
cell antibodies, insulin autoantibodies, and first-phase insulin response in
predicting risk of progression to IDDM in ICA+ relatives: the ICARUS data set. diab 1996;45:1720-1728.
125. Bu DF, Tobin AJ. The exon-intron organization
of the genes (GAD1 and GAD2) encoding two human glutamate decarboxylases (GAD67
and GAD65) suggests that they derive from a common ancestral GAD. Genomics
1994;21(1):222-228.
126. Hagopian WA, Michelsen B, Karlsen AE et al.
Autoantibodies in IDDM primarily recognize the 65,000-M(r) rather than the
67,000-M(r) isoform of glutamic acid decarboxylase. diab 1993;42(4):631-636.
127. Falorni A, Ackefors M, Carlberg C et al.
Diagnostic sensitivity of immunodominant epitopes of glutamic acid
decarboxylase (GAD65) autoantibodies in childhood IDDM. diabetol
1996;39(9):1091-1098.
128. Hallengren B, Falorni A, Landin-Olsson M,
Lernmark A, Papadopoulos KI, Sundkvist G. Islet cell and glutamic acid
decarboxylase antibodies in hyperthyroid patients: at diagnosis and following
treatment. J Intern Med 1996;239(1):63-68.
129. Bottini E, Gerlini G, Lucarini N, Amante A,
Gloria-Bottini F. Evidence of selective interaction between adenosine deaminase
and acid phosphatase polymorphisms in fetuses carried by diabetic women. Hum
Genet 1991;87:199-200.
130. Richter W, Seissler J, Northemann W, Wolfahrt
S, Meinck H-M, Scherbaum WA. Cytoplasmic islet cell antibodies recognize
distinct islet antigens in IDDM but not in stiff man syndrome. diab
1993;42:1642-1648.
131. Syren K, Lindsay L, Stoehrer B et al. Immune
reactivity of diabetes-associated human monoclonal autoantibodies defines
multiple epitopes and detects two domain boundaries in glutamate decarboxylase.
J Immunol 1996;157(11):5208-5214.
132. Daw K, Ujihara N, Atkinson M, Powers AC. Glutamic
acid decarboxylase autoantibodies in Stiff-man syndrome and insulin-dependent
diabetes mellitus exhibit similarities and differences in epitope recognition.
J Immunol 1996;156:818-825.
133. Schwartz HL, Chandonia JM, Kash SF et al.
High-resolution autoreactive epitope mapping and structural modeling of the 65
kDa form of human glutamic acid decarboxylase. J Mol Biol 1999;287(5):983-999.
134. Ronkainen MS, Savola K, Knip M. Antibodies to
GAD65 epitopes at diagnosis and over the first 10 years of clinical type 1
diabetes mellitus. Scand J Immunol 2004;59(3):334-340.
135. Hoppu S, Ronkainen MS, Kulmala P, Akerblom
HK, Knip M. GAD65 antibody isotypes and epitope recognition during the
prediabetic process in siblings of children with type I diabetes. Clin Exp
Immunol 2004;136(1):120-128.
136. Butler MH, Solimena M, Dirkx R, Hayday A, De
Camilli P. Identification of a dominant epitope of glutamic acid decarboxylase
(GAD-65) recognized by autoantibodies in stiff-man syndrome. J Exp Med 1993;178:2097.
137. Kim J, Namchuk M, Bugawan T et al. Higher
autoantibody levels and recognition of a linear NH2-terminal epitope in the
autoantigen GAD65, distinguish stiff-man syndrome from insulin-dependent
diabetes mellitus. J Exp Med 1994;180(2):595-606.
138. Bjork E, Velloso LA, Kampe O, Karlsson FA.
GAD autoantibodies in IDDM, stiff-man syndrome, and autoimmune polyendocrine
syndrome type I recognize different epitopes. Diabetes 1994;43(1):161-165.
139. Powers AC, Bavik K, Tremble J, Daw K,
Scherbaum WA, Banga JP. Comparative analysis of epitope recognition of glutamic
acid decarboxylase (GAD) by autoantibodies from different autoimmune disorders.
Clin Exp Immunol 1999;118(3):349-356.
140. Schories M, Peters T, Rasenack J, Reincke M.
[Autoantibodies against islet cell antigens and type 1 diabetes after treatment
with interferon-alpha]. Dtsch Med Wochenschr 2004;129(20):1120-1124.
141. Schreuder TC, Gelderblom HC, Weegink CJ et
al. High incidence of type 1 diabetes mellitus during or shortly after treatment
with pegylated interferon alpha for chronic hepatitis C virus infection. Liver
Int 2008;28(1):39-46.
142. Li Q, Xu B, Michie SA, Rubins KH, Schreriber
RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese
diabetic mice. Proc Natl Acad Sci U S A 2008;105(34):12439-12444.
143. Fenalti G, Hampe CS, Arafat Y et al.
C-terminal clustering of autoantibody and T cell determinants on the structure
of GAD65 provide insights into the molecular basis of autoreactivity. Diabetes
2008;.
144. Oak S, Gilliam LK, Landin-Olsson M et al. The
lack of anti-idiotypic antibodies, not the presence of the corresponding
autoantibodies to glutamate decarboxylase, defines type 1 diabetes. Proc Natl
Acad Sci U S A 2008;.
145. Hampe CS. Protective role of anti-idiotypic
antibodies in autoimmunity--lessons for type 1 diabetes. Autoimmunity
2012;45(4):320-331.
146. Christie MR, Tun RYM, Lo SSS et al.
Antibodies to GAD and tryptic fragments of islet 64K antigen as distinct
markers for development of IDDM: Studies with identical twins. diab
1992;41:782-787.
147. Christie MR, Genovese S, Cassidy D et al.
Antibodies to islet 37k antigen, but not to glutamate decarboxylase,
discriminate rapid progression to IDDM in endocrine autoimmunity. diab
1994;43(10):1254-1259.
148. Payton MA, Hawkes CJ, Christie MR.
Relationship of the 37,000- and 40,000-Mr tryptic fragments of islet
antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like
molecule IA-2 (ICA512). J Clin Invest 1995;96:1506-1511.
149. Kawasaki E, Yu L, Rewers MJ, Hutton JC,
Eisenbarth GS. Definition of multiple ICA512/phogrin autoantibody epitopes and
detection of intramolecular epitope spreading in relatives of patients with
type 1 diabetes. diab 1998;47:733-742.
150. Gianani R, Rabin DU, Verge CF et al. ICA512
autoantibody radioassay. diab 1995;44(11):1340-1344.
151. Norris JM, Beaty B, Klingensmith G et al.
Lack of association between early exposure to cow's milk protein and b-cell autoimmunity: Diabetes Autoimmunity Study in the Young (DAISY). JAMA
1996;276:609-614.
152. Morran MP, Casu A, Arena VC et al. Humoral
autoimmunity against the extracellular domain of the neuroendocrine autoantigen
IA-2 heightens the risk of type 1 diabetes. Endocrinology 2010;151(6):2528-2537.
153. Farilla
L, Tiberti C, Luzzago A et al. ICA512bdc cDNA expression library displayed on
the capsid protein D of bacteriophage lambda: a novel strategy to identify
major antigenic epitopes in autoimmune diseases. 1999.
Ref Type: Unpublished Work
154. Bonifacio E, Scirpoli M, Kredel K,
Fuchtenbusch M, Ziegler AG. Early autoantibody responses in prediabetes are
IgG1 dominated and suggest antigen-specific regulation. J Immunol
1999;163(1):525-532.
155. Calderon B, Unanue ER. Antigen presentation
events in autoimmune diabetes. Curr Opin Immunol 2012;24(1):119-128.
156. Mohan JF, Levisetti MG, Calderon B, Herzog
JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin
peptides generated within the islets of Langerhans in autoimmune diabetes. Nat
Immunol 2010;11(4):350-354.
157. Stadinski B, Kappler J, Eisenbarth GS.
Molecular targeting of islet autoantigens. Immunity 2010;32(4):446-456.
158. Stadinski BD, Zhang L, Crawford F, Marrack P,
Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7
in an unexpected, weakly binding register. Proc Natl Acad Sci U S A
2010;107(24):10978-10983.
159. Crawford F, Stadinski B, Jin N et al.
Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes
in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A 2011.
160. Nakayama M, Castoe T, Sosinowski T et al.
Germline TRAV5D-4 T-Cell Receptor Sequence Targets a Primary Insulin Peptide of
NOD Mice. diab 2012.
161. Nakayama M, Abiru N, Moriyama H et al. Prime
role for an insulin epitope in the development of type 1 diabetes in NOD mice.
Nature 2005;435(7039):220-223.
162. Pugliese A, Zeller M, Fernandez A et al. The
insulin gene is transcribed in the human thymus and transcription levels
correlate with allelic variation at the INS VNTR-IDDM2 susceptibility locus for
type I diabetes. Nat Genet 1997;15(3):293-297.
163. Pugliese A, Fernandez A, Zeller M et al.
Ectopic transcription of IDDM-associated islet cell autoantigen genes and
monoallelic expression of the insulin gene in human thymus. submitted 1996.
164. Garcia CA, Prabakar KR, Diez J et al.
Dendritic cells in human thymus and periphery display a proinsulin epitope in a
transcription-dependent, capture-independent fashion. J Immunol 2005;175:2111-2122.
165. Vafiadis P, Bennett ST, Todd JA et al.
Insulin expression in human thymus is modulated by INS VNTR alleles at the
IDDM2 locus. Nat Genet 1997;15:289-292.
166. Chentoufi AA, Palumbo M, Polychronakos C.
Proinsulin expression by Hassall's corpuscles in the mouse thymus. diab
2004;53(2):354-359.
167. Palumbo MO, Levi D, Chentoufi AA,
Polychronakos C. Isolation and characterization of proinsulin-producing
medullary thymic epithelial cell clones. Diabetes 2006;55(9):2595-2601.
168. Hanahan D. Peripheral-antigen-expressing
cells in thymic medulla: factors in self- tolerance and autoimmunity. Curr Opin
Immunol 1998;10(6):656-662.
169. Davidson HW, Hutton JC. The
insulin-secretory-granule carboxypeptidase H. Biochem J 1993;245:575-582.
170. Mallone R, Brezar V. To B or not to B:
(anti)bodies of evidence on the crime scene of type 1 diabetes? diab
2011;60(8):2020-2022.
171. Berson SA, Yallow RS. Quantitative aspects of
the reaction between insulin and insulin-binding antibody. J Clin Invest
1959;38:1996-2016.
172. Davidson JK, DeBra DW. Immunologic insulin
resistance. diab 1978;27(3):307-318.
173. Hirata Y, Tominaga M, Ito J, Noguchi A.
Spontaneous hypoglycaemia with insulin autoimmunity in Graves' disease. Ann
Intern Med 1974;81:214-218.
174. Uchigata Y, Hirata Y. Insulin Autoimmune
Syndrome (IAS, Hirata Disease). In: Eisenbarth G, editor. Molecular Mechanisms
of Endocrine and Organ Specific Autoimmunity. Austin, Texas: R.G.Landes;
1999:133-148.
175. Ito Y, Nieda M, Uchigata Y et al. Recognition
of human insulin in the context of HLA-DRB1*0406 products by T cells of insulin
autoimmune syndrome patients and healthy donors. J Immunol
1993;151(10):5770-5776.
176. Ziegler AG, Vardi P, Gross DJ et al.
Production of insulin antibodies by mice rejecting insulin transfected cells. J
Autoimmun 1989;2(3):219-227.
177. Srikanta S, Ricker AT, McCulloch DK, Soeldner
JS, Eisenbarth GS, Palmer JP. Autoimmunity to insulin, beta cell dysfunction,
and development of insulin-dependent diabetes mellitus. diab
1986;35(2):139-142.
178. Castano L, Ziegler AG, Ziegler R, Shoelson S,
Eisenbarth GS. Characterization of insulin autoantibodies in relatives of
patients with type 1 diabetes. diab 1993;42:1202-1209.
179. Tiittanen M, Knip M, Vaarala O. Anti-insulin
activity in IgG-fractions from children with newly-diagnosed type 1 diabetes
and negative for insulin autoantibodies. Autoimmunity 2004;37(1):45-49.
180. Vardi P, Ziegler AG, Matthews JH et al.
Concentration of insulin autoantibodies at onset of type I diabetes. Inverse
log-linear correlation with age. Diab care 1988;11(9):736-739.
181. Pugliese A, Bugawan T, Moromisato R et al.
Two subsets of HLA-DQA1 alleles mark phenotypic variation in levels of insulin
autoantibodies in first degree relatives at risk for insulin-dependent
diabetes. J Clin Invest 1994;93:2447-2452.
182. Williams AJK, Bingley PJ, Bonifacio E, Palmer
JP, Gale EAM. A novel micro-assay for insulin autoantibodies. J Autoimmun
1997;10:473-478.
183. Bilbao JR, Calvo B, Urrutia I, Linares A,
Castaño L. Anti-insulin activity in normal newborn cord-blood serum. diab
1997;46:713-716.
184. Naserke HE, Bonifacio E, Ziegler A-G.
Immunoglobulin G insulin autoantibodies in BABYDIAB offspring appear
postnatally: sensitive early detection using a protein A/G-based radiobinding
assay. J Clin Endocrinol Metab 1999;84(4):1239-1243.
185. Yu L, Robles DT, Abiru N et al. Early
expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence
for early determination of subsequent diabetes. Proc Natl Acad Sci USA
2000;97(4):1701-1706.
186. Hummel M, Bonifacio E, Schmid S, Walter M,
Knopff A, Ziegler AG. Brief communication: Early appearance of islet
autoantibodies predicts childhood type 1 diabetes in offspring of diabetic
parents. Annals of Internal Medicine 2004;140(11):882-886.
187. Siljander H, Harkonen T, Hermann R et al.
Role of insulin autoantibody affinity as a predictive marker for type 1
diabetes in young children with HLA-conferred disease susceptibility. Diabetes
Metab Res Rev 2009;25(7):615-622.
188. Hoppu S, Ronkainen MS, Kimpimaki T et al.
Insulin autoantibody isotypes during the prediabetic process in young children
with increased genetic risk of type 1 diabetes. Pediatr Res 2004;55(2):236-242.
189. Abiru N, Maniatis AK, Yu L et al. Peptide and
MHC specific breaking of humoral tolerance to native insulin with the B:9-23
peptide in diabetes prone and normal mice. diab 2001;50:1274-1281.
190. Moriyama H, Abiru N, Paronen J et al.
Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and
diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A
2003;100(18):10376-10381.
191. Liu E, Moriyama H, Abiru N. et al.
Anti-peptide autoantibodies and fatal anaphylaxis in NOD mice in response to
insulin self-peptides B:9-23 and B:13-23. J Clin Invest 2002;110(7):1021-1027.
192. Liu E, Abiru N, Moriyama H, Miao D,
Eisenbarth GS. Induction of Insulin Autoantibodies and Protection from Diabetes
with Subcutaneous Insulin B:9-23 Peptide Without Adjuvant. Annals New York
Academy of Sciences 2002;958:224-227.
193. Moriyama H, Wen L, Abiru N et al. Induction
and acceleration of insulitis/diabetes in mice with a viral mimic
(polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci
U S A 2002;99(8):5539-5544.
194. Devendra D, Paronen J, Moriyama H, Miao D,
Eisenbarth GS, Liu E. Differential immune response to B:9-23 insulin 1 and
insulin 2 peptides in animal models of type 1 diabetes. Journal of Autoimmunity
2004;23(1):17-26.
195. Yu L, Eisenbarth GS, Bonifacio E, Thomas J,
Atkinson M, Wasserfall C. The Second Murine Autoantibody Workshop: remarkable
inter-laboratory concordance for radiobinding assays to identify insulin
autoantibodies in non-obese diabetic mice. NY Acad Sci 2003;100:1-12.
196. Rojas M, Hulbert C, Thomas JW. Anergy and not
clonal ignorance determines the fate of B cells that recognize a physiological
autoantigen. J Immunol 2001;166(5):3194-3200.
197. Thebault-Baumont K, Dubois-LaForgue D, Krief
P et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD
mice. J Clin Invest 2003;111(6):851-857.
198. Faideau B, Briand JP, Lotton C et al.
Expression of preproinsulin-2 gene shapes the immune response to preproinsulin
in normal mice. J Immunol 2004;172(1):25-33.
199. Kobayashi M, Jasinski J, Liu E et al.
Conserved T cell receptor alpha-chain induces insulin autoantibodies. Proc Natl
Acad Sci U S A 2008;105(29):10090-10094.
200. Wenzlau JM, Frisch LM, Gardner TJ, Sarkar S,
Hutton JC, Davidson HW. Novel antigens in type 1 diabetes: the importance of
ZnT8. Curr Diab Rep 2009;9(2):105-112.
201. Long AE, Gooneratne AT, Rokni S, Williams AJ,
Bingley PJ. The role of autoantibodies to zinc transporter 8 in prediction of
type 1 diabetes in relatives: lessons from the European Nicotinamide Diabetes
Intervention Trial (ENDIT) cohort. J Clin Endocrinol Metab 2012;97(2):632-637.
202. Howson JM, Krause S, Stevens H et al. Genetic
association of zinc transporter 8 (ZnT8) autoantibodies in type 1 diabetes
cases. diabetol 2012;55(7):1978-1984.
203. Wenzlau JM, Moua O, Liu Y, Eisenbarth GS,
Hutton JC, Davidson HW. Identification of a major humoral epitope in Slc30A8
(ZnT8). Ann N Y Acad Sci 2008;1150:252-255.
204. Wenzlau JM, Liu Y, Yu L et al. A common
nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines
ZnT8 autoantibody specificity in type 1 diabetes. diab 2008;57(10):2693-2697.
205. De GJ, Asanghanwa M, Nouthe B et al.
Predictive power of screening for antibodies against insulinoma-associated
protein 2 beta (IA-2beta) and zinc transporter-8 to select first-degree
relatives of type 1 diabetic patients with risk of rapid progression to
clinical onset of the disease: implications for prevention trials. diabetol
2009.
206. Wenzlau JM, Walter M, Gardner TJ et al.
Kinetics of the Post-Onset Decline in Zinc Transporter 8 Autoantibodies in Type
1 Diabetic Human Subjects. J Clin Endocrinol Metab 2010.
207. Vermeulen I, Weets I, Asanghanwa M et al.
Contribution of antibodies against IA-2beta and zinc transporter 8 to
classification of diabetes diagnosed under 40 years of age. Diab care
2011;34(8):1760-1765.
208. Vaziri-Sani F, Oak S, Radtke J et al. ZnT8
autoantibody titers in type 1 diabetes patients decline rapidly after clinical
onset. Autoimmunity 2010.
209. Pietropaolo
M, Babu S, Buelow R, Eisenbarth GS. ICA69: Cloning/sequencing/expression/assay
development. Diabetes 42 (Suppl 1), 4A. 1993.
Ref Type: Abstract
210. Mathews CE, Pietropaolo SL, Pietropaolo M.
Reduced thymic expression of islet antigen contributes to loss of
self-tolerance. Ann N Y Acad Sci 2003;1005:412-417.
211. Stassi G, Schloot N, Pietropaolo M. Islet
cell autoantigen 69 kDa (ICA69) is preferentially expressed in the human islets
of Langerhans than exocrine pancreas. diabetol 1997;40:120-121.
212. Pilon M, Peng XR, Spence AM, Plasterk RH,
Dosch HM. The diabetes autoantigen ICA69 and its Caenorhabditis elegans
homologue, ric-19, are conserved regulators of neuroendocrine secretion. Mol
Biol Cell 2000;11(10):3277-3288.
213. Winer S, Astsaturov I, Gaedigk R et al.
ICA69(null) nonobese diabetic mice develop diabetes, but resist disease
acceleration by cyclophosphamide. J Immunol 2002;168(1):475-482.
214. Karges W, Gaedigk R, Hui MF, Cheung R, Dosch HM.
Molecular cloning of murine ICA69: diabetes-prone mice recognize the human
autoimmune-epitope, Tep69, conserved in splice variants from both species.
Biochim Biophys Acta 1997;1360(2):97-101.
215. Martin S, Kardorf J, Schulte B et al.
Autoantibodies to the islet antigen ICA69 occur in IDDM and in rheumatoid
arthritis. diabetol 1995;38(3):351-355.
216. Atkinson MA, Bowman MA, Kao K et al. Lack of
immune responsiveness to bovine serum albumin in insulin- dependent diabetes. N
Engl J Med 1993;329:1853-1858.
217. Winer S, Gunaratnam L, Astsatourov I et al.
Peptide dose, MHC affinity, and target self-antigen expression are critical for
effective immunotherapy of nonobese diabetic mouse prediabetes. J Immunol
2000;165(7):4086-4094.
218. Fida S, Myers MA, Whittingham S, Rowley MJ,
Ozaki S, Mackay IR. Autoantibodies to the transcriptional factor SOX13 in
primary biliary cirrhosis compared with other diseases. J Autoimmun
2002;19(4):251-257.
219. Davis TME, Mehta Z, Mackay IR et al.
Autoantibodies to the islet cell antigen SOX-13 are associated with duration
but not type of diabetes. Diabetic Med 2003;20(3):198-204.
220. Karounos
DG, Simmerman L, Hickman SL, Jacob RJ. Identification of the p52-Rubella
related autoantigen as an insulin secretory granule protein. Diabetes 42 (Suppl
1), 221A. 1993.
Ref Type: Abstract
221. Honeyman MC, Cram DS, Harrison LC.
Transcription factor jun-B is target of autoreactive T-cells in IDDM. diab
1993;42:626-630.
222. Pak CY, Cha CY, Rajotte RV, McArthur RG, Yoon
JW. Human pancreatic islet cell specific 38 kilodalton autoantigen identified
by cytomegalovirus-induced monoclonal islet cell autoantibody. diabetol
1990;33:569-572.
223. Arden SD, Roep BO, Neophytou PI et al. Imogen
38: a novel 38-kD islet mitochondrial autoantigen recognized by T cells from a
newly diagnosed type I diabetic patient. J Clin Invest 1996;97(2):551-561.
224. Aanstoot HJ, Kang SM, Kim J et al.
Identification and characterization of glima 38, a glycosylated islet cell
membrane antigen, which together with GAD65 and IA2 marks the early phases of
autoimmune response in type 1 diabetes. J Clin Invest 1996;97(12):2772-2783.
225. Winnock F, Christie MR, Batstra MR et al.
Autoantibodies to a 38-kDa glycosylated islet cell membrane-associated antigen
in (pre)type 1 diabetes: association with IA-2 and islet cell autoantibodies.
Diab care 2001;24(7):1181-1186.
226. Saudek CD. When does diabetes start? J A M A
1990;263:2934.
227. Palmen T, Ahola H, Palgi J et al. Nephrin is
expressed in the pancreatic beta cells. Diabetologia 2001;44(10):1274-1280.
228. Yagui K, Shimada F, Mimura M et al. A
missense mutation in the CD38 gene, a novel factor for insulin secretion:
association with Type II diabetes mellitus in Japanese subjects and evidence of
abnormal function when expressed in vitro. diabetol 1998;41(9):1024-1028.
229. Antonelli A, Baj G, Marchetti P et al. Human
anti-CD38 autoantibodies raise intracellular calcium and stimulate insulin
release in human pancreatic islets. diab 2001;50(5):985-991.
230. Park Y, Park H, Yoo E, Kim D. SOX13
autoantibodies are likely to be a supplementary marker for type 1 diabetes in
Korea. Ann N Y Acad Sci 2003;1005:253-258.
231. Kundu SK, Pleatman MA, Redwine WA, Boyd AE,
Marcus DM. Binding of monoclonal antibody A2B5 to gangliosides. Biochem Biophys
Res Commun 1983;116(3):836-842.
232. Nayak RC, Attawia NA, Cahill CJ, King GL,
Ohashi O, Moromisato R. Expression of a monoclonal antibody (3G5) defined
ganglioside antigen in renal cortex. Kidney Int 1992;41:1638-1645.
233. Alejandro R, Shienvold FL, Hajek SAV, Pierce
M, Paul R, Mintz DH. A ganglioside antigen on the rat pancreatic B cell surface
identified by monoclonal antibody R2D6. J Clin Invest 1984;74:25-38.
234. Srikanta S, Krisch K, Eisenbarth GS. Islet cell
proteins defined by monoclonal islet cell antibody HISL-19. diab
1986;35(3):300-305.
235. Srikanta S, Telen M, Posillico JT et al.
Monoclonal antibodies to a human islet cell surface glycoprotein: 4F2 and
LC7-2. Endocrinology 1987;120(6):2240-2244.
236. Gillard BK, Thomas JW, Nell LJ, Marcus DM.
Antibodies against ganglioside GT3 in the sera of patients with type I diabetes
mellitus. J Immunol Methods 1989;142(11):3826-3832.
237. Buschard
K, Josefsen K, Horn T, Larsen S, Fredman P. Sulphatide and anti-sulphatide
antibodies in type I diabetes mellitus. Diabetes 42 (Suppl 1), 5A. 1993.
Ref Type: Abstract
238. Dotta F, Previti M, Lenti L et al. GM2-1
pancreatic islet ganglioside: identification and characterization of a novel
islet-specific molecule. diabetol 1995;38:1117-1121.
239. Dotta
F, Gianani R, Dionisi S, Tiberti C, Eisenbarth GS, DiMario U. Expression of
anti-GM2-1 islet ganglioside antibodies in ICA+ relatives of type I diabetics.
Autoimmunity 15 (suppl), 70. 1993.
Ref Type: Abstract
240. Dotta F, Colman PG, Lombardi D et al.
Ganglioside expression in human pancreatic islets. diab 1989;38(11):1478-1483.
241. Cohen IR. Thoughts on the pathogenesis of
type 1 diabetes and on the arrest of autoimmune beta-cell destruction by
peptide p277 vaccination. Isr Med Assoc J 2004;6(5):260-261.
242. Elias D, Reshef T, Birk OS, Van der Zee R,
Walker MD, Cohen IR. Vaccination against autoimmune mouse diabetes with a
T-cell epitope of the human 65-kDa heat shock protein. Proc Natl Acad Sci USA
1991;88(8):3088-3091.
243. Brugman S, Klatter FA, Visser J, Bos NA,
Elias D, Rozing J. Neonatal oral administration of DiaPep277, combined with
hydrolysed casein diet, protects against Type 1 diabetes in BB-DP rats. An
experimental study. diabetol 2004;47(7):1331-1333.
244. Bowman M, Atkinson MA. Heat shock protein
therapy fails to prevent diabetes in NOD mice. diabetol 2002;45(9):1350-1351.
245. Jones DB, Hunter NR, Duff GW. Heat-shock
protein 65 as a B cell antigen of insulin-dependent diabetes. Lancet 1990;336:583-585.
246. Horvath L, Cervenak L, Oroszlan M et al.
Antibodies against different epitopes of heat-shock protein 60 in children with
type 1 diabetes mellitus. Immunol Lett 2002;80(3):155-162.
247. Elias D, Meilin A, Ablamunits V et al. Hsp60 peptide
therapy of NOD mouse diabetes induces a Th2 cytokine burst and downregulates
autoimmunity to various b-cell antigens. diab
1997;46:758-764.
248. Inman LR, McAllister CT, Chen L et al.
Autoantibodies to the GLUT-2 glucose transporter of b cells in insulin-dependent diabetes mellitus of recent onset. Proc Natl
Acad Sci USA 1993;90:1281-1284.
249. Johnson JH, Crider BP, McCorkle K, Alford M,
Unger RH. Inhibition of glucose transport into rat islet cells by
immunoglobulins from patients with new-onset insulin-dependent diabetes
mellitus. N Engl J Med 1990;322:653-659.
250. Shehadeh N, Pollack S, Wildbaum G et al.
Selective autoantibody production against CCL3 is associated with human type 1
diabetes mellitus and serves as a novel biomarker for its diagnosis. J Immunol
2009;182(12):8104-8109.
251. Gurr W, Shaw M, Li Y, Sherwin R. RegII is a
beta-cell protein and autoantigen in diabetes of NOD mice. diab
2007;56(1):34-40.
252. Gurr W, Yavari R, Wen L et al. A Reg family
protein is overexpressed in islets from a patient with new-onset type 1
diabetes and acts as T-cell autoantigen in NOD mice. diab 2002;51(2):339-346.
253. Astorri E, Guglielmi C, Bombardieri M et al.
Circulating Reg1alpha proteins and autoantibodies to Reg1alpha proteins as
biomarkers of beta-cell regeneration and damage in type 1 diabetes. Horm Metab
Res 2010;42(13):955-960.
254. Oling V, Reijonen H, Simell O, Knip M, Ilonen
J. Autoantigen-specific memory CD4+ T cells are prevalent early in progression
to Type 1 diabetes. Cell Immunol 2012;273(2):133-139.
255. Pugliese A, Reijonen HK, Nepom J, Burke GW,
III. Recurrence of autoimmunity in pancreas transplant patients: research
update. Diabetes Manag (Lond) 2011;1(2):229-238.
256. Standifer NE, Burwell EA, Gersuk VH,
Greenbaum CJ, Nepom GT. Changes in autoreactive T cell avidity during type 1
diabetes development. Clin Immunol 2009;132(3):312-320.
257. Coppieters KT, Dotta F, Amirian N et al.
Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from
recent onset and long-term type 1 diabetes patients. J Exp Med 2012.
258. Tsai S, Shameli A, Santamaria P. CD8+ T cells
in type 1 diabetes. Adv Immunol 2008;100:79-124.
259. Medarova Z, Tsai S, Evgenov N, Santamaria P,
Moore A. In vivo imaging of a diabetogenic CD8+ T cell response during type 1
diabetes progression. Magn Reson Med 2008;59(4):712-720.
260. Lieberman SM, Evans AM, Han B et al.
Identification of the {beta} cell antigen targeted by a prevalent population of
pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A
2003;100(14):8384-8388.
261. Hutton JC, Eisenbarth GS. A pancreatic
{beta}-cell-specific homolog of glucose-6-phosphatase emerges as a major target
of cell-mediated autoimmunity in diabetes. Proc Natl Acad Sci U S A
2003;100:8626-8628.
262. Santamaria P, Utsugi T, Park B-J, Averill N,
Kawazu S, Yoon J-W. Beta-cell-cytotoxic CD8+ T cells from nonobese diabetic
mice use highly homologous T cell receptor a-chain CDR3
sequences. J Immunol 1995;154:2494-2503.
263. Trudeau JD, Kelly-Smith C, Verchere CB et al.
Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of
autoreactive T cells in peripheral blood. J Clin Invest 2003;111(2):217-223.
264. Qin H, Trudeau JD, Reid GS et al. Progression
of spontaneous autoimmune diabetes is associated with a switch in the killing
mechanism used by autoreactive CTL. Int Immunol 2004;16(11):1657-1662.
265. Oeser JK, Parekh VV, Wang Y et al. Deletion
of the G6pc2 Gene Encoding the Islet-Specific Glucose-6-Phosphatase Catalytic
Subunit-Related Protein Does Not Affect the Progression or Incidence of Type 1
Diabetes in NOD/ShiLtJ Mice. diab 2011.
266. Krishnamurthy B, Mariana L, Gellert SA et al.
Autoimmunity to Both Proinsulin and IGRP Is Required for Diabetes in Nonobese
Diabetic 8.3 TCR Transgenic Mice. J Immunol 2008;180(7):4458-4464.
267. Krishnamurthy B, Dudek NL, McKenzie MD et al.
Responses against islet antigens in NOD mice are prevented by tolerance to
proinsulin but not IGRP. J Clin Invest 2006;116(12):3258-3265.
268. Clemente-Casares X, Tsai S, Huang C,
Santamaria P. Antigen-specific therapeutic approaches in type 1 diabetes. Cold
Spring Harb Perspect Med 2012;2(2):a007773.
269. Krishnamurthy B, Chee J, Jhala G et al.
Complete diabetes protection despite delayed thymic tolerance in NOD8.3 TCR
transgenic mice due to antigen-induced extrathymic deletion of T cells. diab
2012;61(2):425-435.
270. Fisher LD, Kennedy JW, Davis KB et al.
Association of sex, physical size, and operative mortality after coronary
artery bypass in the Coronary Artery Surgery Study (CASS). J Thorac Cardiovasc
Surg 1982;84:334-341.
271. Wong FS, Siew LK, Scott G et al. Activation
of insulin-reactive CD8 T-cells for development of autoimmune diabetes.
Diabetes 2009;58(5):1156-1164.
272. Skowera A, Ellis RJ, Varela-Calvino R et al.
CTLs are targeted to kill beta cells in patients with type 1 diabetes through
recognition of a glucose-regulated preproinsulin epitope. J Clin Invest
2008;118(10):3390-3402.
273. Velthuis JH, Unger WW, Abreu JR et al.
Simultaneous detection of circulating autoreactive CD8+ T-cells specific for
different islet cell-associated epitopes using combinatorial MHC-multimers.
diab 2010.
274. Wegmann DR, Norbury-Glaser M, Daniel D.
Insulin-specific T cells are a predominant component of islet infiltrates in
pre-diabetic NOD mice. Eur J Immunol 1994;24(8):1853-1857.
275. Daniel D, Gill RG, Schloot N, Wegmann D.
Epitope specificity, cytokine production profile and diabetogenic activity of
insulin-specific T cell clones isolated from NOD mice. Eur J Immunol
1995;25(4):1056-1062.
276. Daniel D, Wegmann DR. Protection of nonobese
diabetic mice from diabetes by intranasal or subcutaneous administration of
insulin peptide B-(9-23). Proc Natl Acad Sci USA 1996;93(2):956-960.
277. Abiru N, Wegmann D, Kawasaki E, Gottlieb P,
Simone E, Eisenbarth GS. Dual overlapping peptides recognized by insulin
peptide B:9-23 reactive T cell receptor AV13S3 T cell clones of the NOD mouse.
J Autoimmun 2000;14(3):231-237.
278. Simone E, Daniel D, Schloot N et al. T cell
receptor restriction of diabetogenic autoimmune NOD T cells. Proc Natl Acad Sci
USA 1997;94(6):2518-2521.
279. Zhang L, Jasinski JM, Kobayashi M et al.
Analysis of T cell receptor beta chains that combine with dominant conserved
TRAV5D-4*04 anti-insulin B:9-23 alpha chains. J Autoimmun 2009.
280. Alleva DG, Crowe PD, Jin L et al. A
disease-associated cellular immune response in type 1 diabetics to an
immunodominant epitope of insulin. J Clin Invest 2001;107(2):173-180.
281. Reunanen A, Akerblom HK, Kaar ML. Prevalence
and ten-year (1970-1979) incidence of insulin- dependent diabetes mellitus in
children and adolescents in Finland. Acta Paediatr Scand 1982;71(6):893-899.
282. Daniel C, Weigmann B, Bronson R, von BH.
Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong
agonist insulin mimetope. J Exp Med 2011;208(7):1501-1510.
283. Kent SC, Chen Y, Bregoli L et al. Expanded T
cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an
insulin epitope. Nature 2005;435(7039):224-228.
284. Wong FS, Karttunen J, Dumont C et al.
Identification of an MHC class I-restricted autoantigen in type 1 diabetes by
screening an organ-specific cDNA library. Nat Med 1999;5(9):1026-1031.
285. Chen W, Bergerot I, Elliott JF et al.
Evidence that a peptide spanning the B-C junction of proinsulin is an early
autoantigen epitope in the pathogenesis of type 1 diabetes. J Immunol 2001;167(9):4926-4935.
286. Halbout P, Briand JP, Becourt C, Muller S,
Boitard C. T cell response to preproinsulin I and II in the nonobese diabetic
mouse. J Immunol 2002;169(5):2436-2443.
287. Durinovic-Bello I, I, Boehm BO, Ziegler AG.
Predominantly Recognized ProInsulin T Helper Cell Epitopes in Individuals With
and Without Islet Cell Autoimmunity. J Autoimmun 2002;18(1):55-66.
288. Narendran P, Mannering SI, Harrison LC.
Proinsulin - a pathogenic autoantigen in type 1 diabetes. Autoimmunity Reviews
2003;2(4):204-210.
289. Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S,
Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and
phenotype of the autoimmune response to proinsulin. Genes Immun 2010.
290. Haskins K. Pathogenic T-cell clones in
autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol
2005;87:123-62.:123-162.
291. Dobbs C, Haskins K. Comparison of a T cell
clone and of T cells from a TCR transgenic mouse: TCR transgenic T cells
specific for self-antigen are atypical. J Immunol 2001;166(4):2495-2504.
292. Hoglund P, Mintern J, Waltzinger C, Heath W,
Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally
regulated presentation of islet cell antigens in the pancreatic lymph nodes. J
Exp Med 1999;189(2):331-339.
293. Andre-Schmutz I, Hindelang C, Benoist C,
Mathis D. Cellular and molecular changes accompanying the progression from
insulitis to diabetes. Eur J Immunol 1999;29(1):245-255.
294. Stadinski BD, Delong T, Reisdorph N et al.
Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol
2010;11(3):225-231.
295. Nikoopour E, Sandrock C, Huszarik K et al.
Cutting Edge: Vasostatin-1-Derived Peptide ChgA29-42 Is an Antigenic Epitope of
Diabetogenic BDC2.5 T Cells in Nonobese Diabetic Mice. J Immunol
2011;186(7):3831-3835.
296. Gurr W, Yavari R, Wen L et al. A Reg family
protein is overexpressed in islets from a patient with new-onset type 1
diabetes and acts as T-cell autoantigen in NOD mice. diab 2002;51(2):339-346.
297. Atkinson MA, Kaufman DL, Campbell L et al.
Response of peripheral-blood mononuclear cells to glutamate decarboxylase in
insulin-dependent diabetes. Lancet 1992;339(8791):458-459.
298. Atkinson MA, Bowman MA, Campbell L, Darrow
BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to
glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J
Clin Invest 1994;94(5):2125-2129.
299. Herskowitz R, Jackson RA. Pilot trial of
preventive therapy: progression to overt hyper- glycemia by 3/3
"prediabetics" despite oral nicotinamide. diab 1988;37:59A.
300. Mannering SI, Morris JS, Jensen KP et al. A
sensitive method for detecting proliferation of rare autoantigen-specific human
T cells. J Immunol Methods 2003;283(1-2):173-183.
301. Van der Auwera B, Van Waeyenberge C, Schuit F
et al. DRB1*0403 protects against IDDM in Caucasians with the high-risk
heterozygous DQA1*0301-DQB1*0302/DQA1*501-DQB1*0201 genotype. diab
1995;44:527-530.
302. Tisch R, Yang X-D, Singer SM, Liblau RS,
Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase
correlates with insulitis in non-obese diabetic mice. Nature 1993;366:72-75.
303. Tisch R, Liblau RS, Yang X-D, Liblau P,
McDevitt HO. Induction of GAD65-specific regulatory T-cells inhibits ongoing
autoimmune diabetes in nonobese diabetic mice. diab 1998;47:894-899.
304. Mallone R, Kochik SA, Laughlin EM et al.
Differential Recognition and Activation Thresholds in Human Autoreactive
GAD-Specific T-Cells. diab 2004;53(4):971-977.
305. Nepom GT, Lippolis JD, White FM et al.
Identification and modulation of a naturally processed T cell epitope from the
diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65).
Proc Natl Acad Sci U S A 2001;98(4):1763-1768.
306. Laughlin E, Burke G, Pugliese A, Falk B,
Nepom G. Recurrence of autoreactive antigen-specific CD4+ T cells in autoimmune
diabetes after pancreas transplantation. Clin Immunol 2008;128(1):23-30.
307. Roep BO, Atkinson MA, Van Endert PM, Gottlieb
PA, Wilson SB, Sachs JA. Autoreactive T cell Responses in Insulin-dependent
(Type 1) Diabetes Mellitus. Report of the First International Workshop for
Standardization of T cell assays. J Autoimmun 1999;13(2):267-282.
308. Schloot NC, Meierhoff G, Karlsson FM et al.
Comparison of cytokine ELISpot assay formats for the detection of islet antigen
autoreactive T cells. Report of the third immunology of diabetes society T-cell
workshop. J Autoimmun 2003;21(4):365-376.
309. Pinkse GG, Boitard C, Tree TI, Peakman M,
Roep BO. HLA class I epitope discovery in type 1 diabetes: independent and
reproducible identification of proinsulin epitopes of CD8 T cells--report of
the IDS T Cell Workshop Committee. Ann N Y Acad Sci 2006;1079:19-23.
310. Di Lorenzo TP, Peakman M, Roep BO. Translational
mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes
in autoimmune diabetes. Clin Exp Immunol 2007;148(1):1-16.
311. Seyfert-Margolis V, Gisler TD, Asare AL et
al. Analysis of T-cell assays to measure autoimmune responses in subjects with
type 1 diabetes: results of a blinded controlled study. Diabetes
2006;55(9):2588-2594.
312. Gianani R, Putnam A, Still T et al. Initial
results of screening of non - diabetic organ donors for expression of islet
autoantibodies. J Clin Endocrinol Metab 2006;91:1855-1861.
313. Maniatis AK, Yu L, Miao D, Nelson K,
Eisenbarth GS. Rapid Assays for Detection of Anti-islet Autoantibodies:
Implications for Organ Donor Screening. J Autoimmun 2001;16(1):71-76.
314. Gianani R, Campbell-Thompson M, Sarkar SA et
al. Dimorphic histopathology of long-standing childhood-onset diabetes.
diabetol 2010.
315. Bosi E, Braghi S, Maffi P et al. Autoantibody
response to islet transplantation in type 1 diabetes. diab
2001;50(11):2464-2471.
316. Shapiro AM, Ricordi C, Hering BJ et al.
International trial of the Edmonton protocol for islet transplantation. N Engl
J Med 2006;355(13):1318-1330.
317. Vendrame F, Pileggi A, Laughlin E et al.
Recurrence of Type 1 Diabetes after Simultaneous Pancreas-Kidney Transplantation,
despite Immunosuppression, Associated with Autoantibodies and Pathogenic
Autoreactive CD4 T-cells. diab 2010.
318. Bingley PJ, Bonifacio E, Williams AJK,
Genovese S, Bottazzo GF, Gale EAM. Prediction of IDDM in the general
population: strategies based on combinations of autoantibody markers. diab
1997;46:1701-1710.
319. Maclaren N, Lan M, Coutant R et al. Only
multiple autoantibodies to islet cells (ICA), insulin, GAD65, IA-2 and IA-2beta
predict immune-mediated (Type 1) diabetes in relatives. J Autoimmun
1999;12(4):279-287.
320. Knip M, Korhonen S, Kulmala P et al.
Prediction of type 1 diabetes in the general population. Diab care
2010;33(6):1206-1212.
321. Hummel M, Ziegler AG, Roth R. Psychological
impact of childhood islet autoantibody testing in families participating in the
BABYDIAB study. Diabet Med 2004;21(4):324-328.
322. Carmichael SK, Johnson SB, Baughcum A et al.
Prospective assessment in newborns of diabetes autoimmunity (PANDA): maternal
understanding of infant diabetes risk. Genet Med 2003;5(2):77-83.
323. Gustafsson SU, Ludvigsson J, Liss PE,
Svensson T. Bioethical theory and practice in genetic screening for type 1
diabetes. Med Health Care Philos 2003;6(1):45-50.
324. Yu MS, Norris JM, Mitchell CM et al. Impact
on maternal parenting stress of receipt of genetic information regarding risk
of diabetes in newborn infants. Am J Med Genet 1999;86(3):219-226.
325. Hahl J, Simell T, Ilonen J, Knip M, Simell O.
Costs of predicting IDDM. diabetol 1998;41:79-85.
326. Barker
JM, Goehrig SH, Barriga K et al. Clinical characteristics of children diagnosed
with type 1 diabetes through intensive screening and follow-up. Diab care
2004;27(6):1399-1404.