Chapter 11
Prediction of Type 1A Diabetes:
The Natural History of the Prediabetic
Period
Updated 09-2012
George S. Eisenbarth
Introduction
Predictive Factors in Relatives
Prediction in the General Population
Prediction/Diagnosis in Adults
Stage in Life Initiation
Are there Abnormalities that Precede Autoantibodies?
Environmental Factors?
Are Beta Cells Destroyed in a Progressive/Linear Fashion?
Conclusions
Introduction
The importance of understanding the natural history of immune mediated pre-diabetes1-3 lies in the development of prevention strategies4-6. Several initial randomized clinical intervention trials have concluded and the next generation of such trials will rely upon improved and simplified identification of individuals7 who are at high risk of progression to diabetes8. This is essential to ensure that trials will have sufficient statistical power to detect a given effect of the intervention (if it exists) within the time available for the study. Such understanding is also needed to avoid exposing those who will not develop diabetes to the risk of adverse effects of the intervention. In addition, it is likely that many interventions will be more effective if given early, with more extant beta cells. In addition there is accumulating evidence that at the onset of type 1A diabetes, and in a subset of patients years after the onset of diabetes (Figure 11.1) there remains islet beta cells9, and preservation of even low levels of insulin secretion has multiple benefits in terms of improved glycemic control and prevention of complications10-14.
The amount of beta cell destruction at the onset of diabetes remains an open question15-20 with one estimate that a 40% reduction in beta cell mass is sufficient for diabetes of a 20 year old while 80-90% loss may be required for children presenting with diabetes less than age 521. Given individual differences in rates of progression to diabetes and insulin resistance (as well as potential function per beta cell), it is likely that the variance of beta cell loss will be large at any age. It is of interest that the slope of loss of c-peptide after diabetes onset decreased modestly from the immediate prediabetic phase22. This may reflect “synchronization” with greater rate of loss associated with presentation with diabetes. With long-term type 1 diabetes the mean beta cell loss is dramatic(Figure 10.1).
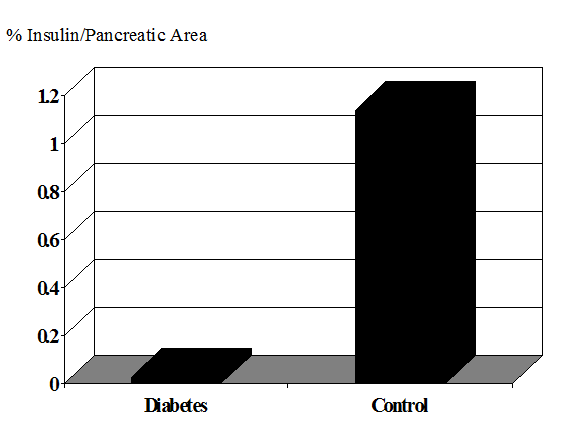
Figure 11.1.
Area of insulin containing cells in long-term patients with diabetes 23.
First-degree relatives of individuals with type 1A diabetes have an approximate 5% risk of developing the disease (independent of country 24) while children without a relative with diabetes in the United States have a risk of 1/300 while in Japan the risk is less than 1/3,000. Longitudinal studies of autoantibody-positive relatives and more recently general populations25, 26 have provided a wealth of information on the natural history of autoimmunity during the pre-hyperglycemic phase of the disease (prediabetes). These studies have established the predictive value of age27, islet cell antibodies (ICAs)28, multiple “biochemical” autoantibodies 29-32 first phase insulin release (FPIR)33, impaired glucose tolerance, C-peptide secretion (Prediction”Scores”) 34, and human leukocyte (HLA) haplotypes35. Increasingly, combinations of markers are being used to better define the risk of diabetes 36-40 Prediction is not absolute, but can be expressed as the percentage of individuals developing diabetes within a given time period. In addition the age at which islet autoantibodies first appear and levels of insulin autoantibodies (not antibodies reacting with GAD, IA-2 or ZnT8) predict approximate age of onset of type 1 diabetes25, 26. This chapter will review predictive factors currently in use and discuss some of the unanswered questions on the natural history of Type 1A (immune mediated) prediabetes.
Predictive Factors in Relatives
The first large
scale studies of the prediction of type 1A diabetes relied upon the detection
of cytoplasmic islet cell autoantibodies (
Riley and
coworkers reported on the
The
Bart's-Windsor family study, conducted in
Figure 11.2
Data from the Joslin Diabetes Center family study indicate that impaired FPIR (First Phase Insulin Release, usually analyzed as the sum of insulin at 1 and 3 minutes following a bolus of intravenous glucose) is an additional risk factor33. Thirty-five first-degree relatives with high-titer ICAs (> 40 JDF units) underwent serial intravenous glucose tolerance testing (IVGTT). The age of the subjects ranged from 2.6 to 66 years, the mean follow-up was 3.6 years from the first test, and 18 progressed to overt diabetes. The FPIR was calculated as the sum of the 1- and 3-minute insulin levels after a standard bolus of intravenous glucose (0.5 g per kg body weight, infused as a 20%-25% solution over 2 to 4 minutes). Percentiles for the FPIR were determined in 225 healthy, non-obese, control subjects. Even in control subjects the FPIR showed wide within-subject variation (discussed in detail below). Nevertheless, relatives with an initial FPIR below the first percentile (48 mU/l) had significantly reduced diabetes-free survival. Importantly, the presence of FPIR below the first percentile did not signify that diabetes was already present47-50. For most of the relatives, oral glucose tolerance tests (OGTT) were also performed during follow-up to detect asymptomatic diabetes. For the survival analysis the onset of diabetes was defined by a diabetic OGTT or the occurrence of symptomatic hyperglycemia, or whichever came first. A number of recent studies are analyzing impaired fasting glucose, impaired glucose tolerance (2 hour glucose on oral glucose tolerance testing)51, c-peptide secretion52 and potential correlates of insulin resistance (e.g. HOMA-R) and there is evidence of abnormalities preceding diabetes even in the subset of individuals with relatively normal first phase insulin secretion53, 54. The average time from the discovery FPIR below the first percentile to the onset of diabetes was 1.8 years. Sosenko and coworkers for DPT and Trialnet data have developed risk scores that primarily depend on quantitation of glucose tolerance testing. Though there is variability between subsequent tests, in general, as glucose increases risk increases especially in children53, 54.
Updated data
from the Joslin family study, with longer follow-up and larger numbers of
relatives, have confirmed these findings.
Among 79 relatives with high titer ICAs (> 40 JDF units),
those with FPIR below the first percentile on the first test had 3-year
diabetes-free survival of 13% (95% confidence interval: 0%-30%) compared with a
78% (95% confidence interval: 63%-93%) for the group with higher FPIR55. Studies at the Barbara Davis Center have
confirmed the predictive value of FPIR measurements as has studies from the
Melbourne family study, studies from Finland 56,
and the DPT-1 (Diabetes Prevention Trial)
North American study 57
and analysis of the combined ICARUS database58.
Data from the
Joslin study also suggest that IAA add to the prediction of type 1 diabetes,
but with a weaker effect than
Reports that
identifying ICA subtypes improves the predictive value of ICA can now be put in
the general context of the rule that multiple biochemical autoantibodies are
associated with high risk36. The restricted ICA subtype (reacting with
human and rat islets but not mouse [mouse islets express little or no GAD65]
and with antibody staining restricted to beta cells of rat islets), defined
according to the pattern of sustaining on pancreatic sections, confers a
significantly lower risk of progression to diabetes than a non-restricted
subtype60. Preabsorption of sera with glutamate
decarboxylase (GAD) blocks the ICA staining of restricted ICA-positive sera61 and reduces the ICA staining
of most non-restricted ICA-positive sera62. This suggests that restricted
The ICA assay
is difficult to standardize, is labor intensive, and requires human pancreas36. At the Barbara Davis Center, we utilize a
combination of four assays employing recombinant antigens (insulin
autoantibodies, anti-GAD, anti-ICA512(IA-2) and anti-ZnT8 63) and no longer determine
cytoplasmic ICA. For children the
Genetic factors
can also be considered in assessing diabetes risk64-68. Deschamps and coworkers examined the
predictive value of HLA typing in a study of 536 siblings of diabetic probands
in France69. The risk of type 1 diabetes after 8 years,
estimated by life table analysis, was 10% for siblings who were HLA identical
with the probands, 3%-4% for siblings with either DR3 or DR4, and 16% for those
with DR3/DR4. This compares with 56% for
those with
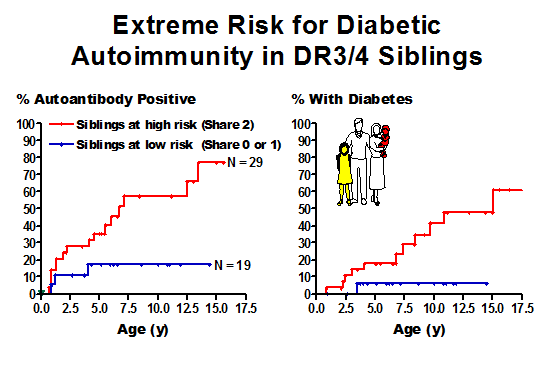
Aly et al Extreme genetic risk for type 1A diabetes67
Figure 11.3 Highest risk
siblings in the DAISY study with DR3/4-DQ2/8 genotype progressing to expression
of islet autoantibodies (left panel) and diabetes (right panel).
In other studies, molecular typing has revealed that the HLA haplotype DQA1*0102 DQB1*0602 confers strong protection from type 1 diabetes, in a dominant fashion (Chapter 7). In our experience, autoantibody-positive relatives with this haplotype have a very low risk of progression to diabetes71 and usually express only a single autoantibody, namely anti-GAD, although a few also express IAA. Such protection is however not absolute, and approximately 1% of children developing type 1A diabetes72 (versus 20% of the general U.S. population) and 3% of adults with type 1 diabetes have DQB1*0602 (DQB1*0602 is usually part of the haplotype DRB1*1501, DQA1*0102, DQB1*0602). Approximately 5% of older individuals developing type 1 diabetes are reported to have the protective HLA allele DQB1*060273.
In addition to HLA more than 50 loci contribute to risk of type 1 diabetes74. Each locus has a small effect but a report by Winkler and coworkers suggests that combining loci can impact prediction68. We have followed a set of identical triplets and now all three triplets have progressed to diabetes including the last triplet who was non-diabetic in 1983 (Figure 11.4).
Prediction in the General Population
It is likely
that genetic typing will have an even greater impact on assessing diabetes risk
in the general population25, 68, 75, 76. Most studies of prediabetic subjects have
involved the screening of first-degree relatives of diabetic probands, rather
than the general population. However,
less than 10% of new cases of type 1 diabetes have an affected relative, so the
general population will need to be screened eventually if an effective
intervention is to have a major impact.
Screening the general population is likely to be more difficult than
screening relatives. Bayes' theorem
states that a screening test will have a lower positive predictive value in the
general population than in a selected group with a higher prevalence of
disease, such as first-degree relatives.
One approach toward solving this problem is to screen the general
population with markers of genetic susceptibility first, followed by
autoantibody testing of susceptible individuals. For example, among the general
Unexpectedly,
studies performed in Florida suggest that ICA have a predictive value in the
general population similar to that in relatives77. In contrast, studies in
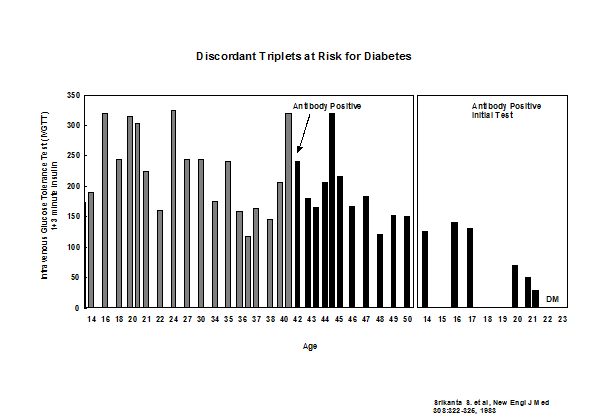
Figure 11.4: Development of autoantibodies and loss of
first phase insulin secretion in identical triplets of a patient with type 1A
diabetes.
Several studies
have now been initiated where children are followed from birth for the
development of anti-islet autoantibodies.
The three studies with the longest follow-up are the BabyDiab study from
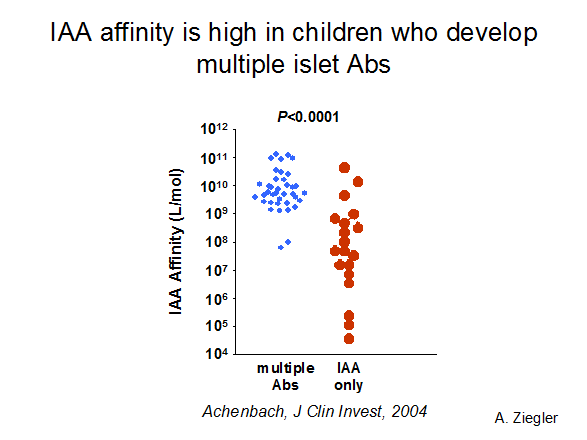
Figure 11.5
Affinity of insulin autoantibodies of children in the BABY-DIAB study,
with high affinity autoantibodies associated with development of multiple islet
autoantibodies and progression to diabetes.
It is likely that there is no age at which a genetically susceptible individual has no increased risk of converting to anti-islet autoantibody positivity89, but progression to diabetes is usually less rapid in older individuals. Figure 11.4 represents updated follow up of a set of non-diabetic monozygotic triplets of a patient with type 1 diabetes, where a second triplet progressed to diabetes at age 21, and at 42 years of age the remaining non-diabetic triplet developed anti-islet autoantibodies and has now progressed to diabetes at age 57. A recent publication updates our studies of monozygotic twins with very high concordance for anti-islet autoimmunity (>60%) given long-term follow up of initially discordant identical twins90 In addition the age of onset of the proband twin and the HLA DR/DQ genotype of the twins contribute to differences in long term risk (higher cumulative risk the younger the age of onset of the proband and higher risk with the DR3/-DQ2/8 genotype).
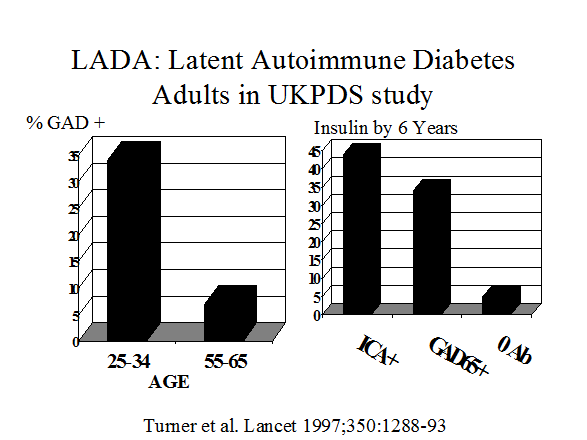
Figure 11.6
Prediction/Diagnosis in Adults
Both patients with gestational diabetes (Figure 11.7) and adults with a diagnosis of type 2 diabetes (Figure 11.6)91 have a significant risk of having type 1A diabetes92, 93. Between 5 and 10% of patients with a diagnosis of gestational diabetes express anti-islet autoantibodies and the great majority progress to type 1A diabetes. In a similar manner, the diagnosis of type 1 diabetes is associated with expression of islet autoantibodies94 in adults thought initially to have type 2 diabetes95, 96, 96. Assays for GAD65 autoantibodies are the most useful for type 1A diabetes in both patients with gestational or adult onset type 1 diabetes97-99 and the term LADA (Latent Autoimmunity of Adults) has been applied to latter this group. 100 In addition to autoantibody assays specialized assays for detection of T cells reacting with islet autoantigens may identify a subset of patients with a clinical diagnosis of Type 2 diabetes who have anti-islet autoimmunity.101, 102 At present such T cell assays are performed in relatively few laboratories without workshop standardization.
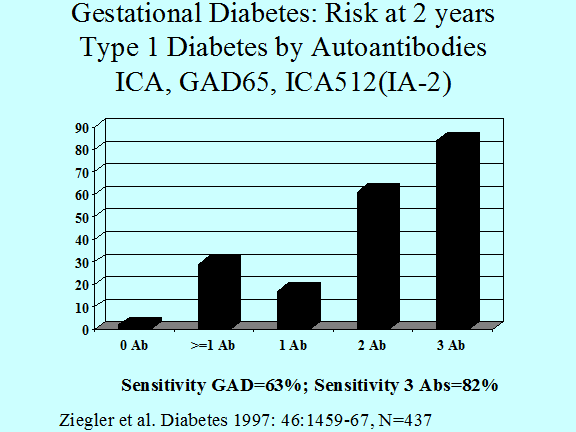
Figure 11.7 Progression to insulin dependent
diabetes of individuals with gestational diabetes (diabetes developing during
pregnancy) analyzed relative to the number of anti-islet autoantibodies.
At What Stage in Life is Beta Cell Autoimmunity Initiated, What Is the
Initial Target Antigen, and in What Sequence Do Different Autoantibodies
Appear?
Studies in first-degree relatives have detected evidence of beta cell autoimmunity many years before clinical presentation with overt hyperglycemia27, 28, 103. However, the existence of young infants with type 1A diabetes indicates that beta cell destruction may also occur rapidly in some individuals. A plausible hypothesis is that beta cell autoimmunity can begin early in life, with variation in the age of onset of overt diabetes explained by differences in the rate of beta cell destruction from one individual to another, but there is also extreme variation in the age which autoantibodies first develop. Thus both the timing of appearance of islet autoantibodies and the rate of progression once autoantibodies are detected are associated with age of onset25. The first autoantibody to appear is usually insulin autoantibodies, followed by GAD65 autoantibodies (Figure 11.8) but this is a generalization, as is the observation that IA-2 and ZnT8 autoantibodies follow after several years. Any of the autoantibodies can be the first or the only autoantibody expressed. There is a misconception that the levels of insulin autoantibodies are determined by the age of the individual. Though levels of insulin autoantibodies are much higher in children developing type 1 diabetes with onset less than age 5104, when prediabetic children are followed from birth the levels are not related to age but the rate of progression to diabetes25. Figure 11.9 illustrates a child in the DAISY study followed from birth with 13 years of autoantibody positivity before progression to diabetes. Such long prodromes are associated with either none or low levels of insulin autoantibodies (bottom line) even at one year of age.
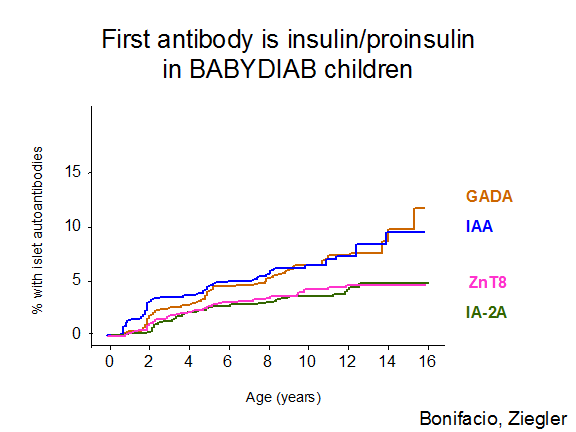
Figure 11.8 Cumulative development of islet autoantibodies in the BabyDiab study 2009).
In support of the hypothesis that the timing of diabetes onset is influenced by factors such as insulin resistance, a major peak in the incidence of type 1A diabetes occurs during early adolescence in nearly all populations studied105. This increased incidence during adolescence is probably due to the insulin resistance associated with puberty, with individuals requiring more insulin than can be produced given beta cell destruction.
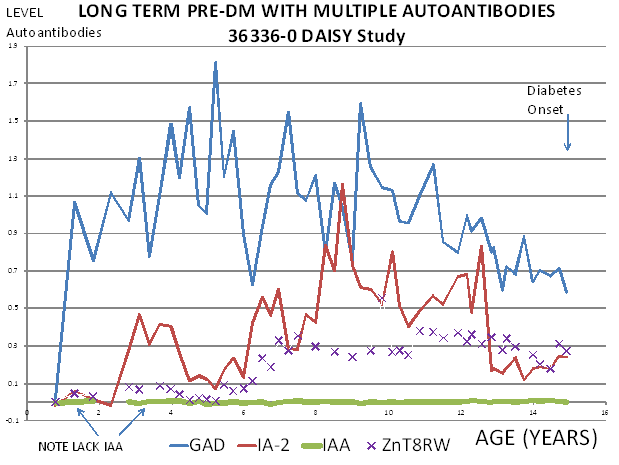
Figure 11.9 Child with long
prodrome preceding diabetes with typical lack of insulin autoantibodies
throughout follow up
After the age
of about 15 years, measuring the incidence of type 1A diabetes is complicated
by misclassification of some patients as type II diabetics. Nevertheless, it has been estimated that at
least 37% of type 1 diabetes occurs after the age of 19 years and 15% after the
age of 30 years106. Adult autoimmune diabetes differs relatively
little genetically from pediatric diabetes107. It has been suggested that there is an excess
of type 2 diabetes in parents of children with type 1A diabetes, but a report
of the analysis of parents in the Barts-Windsor study indicates that the
majority of diabetic parents have type 1A diabetes, and the prevalence of type
2 diabetes is not higher compared to the general population108.
Table 11.1
|
Country |
Age Range |
Number Screened |
Prevalence of
|
Cutoff (JDF
Units) |
Incidence of
Type 1 Diabetes (a) |
References |
|
|
7-18 |
473 |
0.4% |
-- |
2.0 |
109, 110 |
|
|
6-21 |
4287 |
1.05% |
>/=5 |
7.0 |
111, 112 |
|
|
|
|
0.4% |
>/=20 |
|
|
|
|
6-17 |
8363 |
1.79% |
>/=4.5 |
7.1 |
113, 114 |
|
|
|
|
0.2% |
>/=24 |
|
|
|
|
5-19 |
4806 |
0.42% |
>/=.06 |
11.0 |
115, 116 |
|
|
14-17 |
2291 |
0.35% |
>/=5 |
11.3 |
117, 118 |
|
|
5-7 |
20000 |
1.2% |
>/=10 |
12.8 |
119, 120 |
|
|
|
|
|
|
|
|
|
|
5-18 |
9696 |
0.59% |
>/=10 |
13.7(b) |
77 |
|
State |
12-18 |
3992 |
1.7% |
>/=1 |
|
121 |
|
Pennsylvan |
|
|
|
|
|
122 |
|
|
4-18 |
1900 |
0.68% |
>/=20 |
14.5 |
123, 124 |
|
|
9-13 |
2925 |
2.8% |
>/=4 |
15.6 |
78, 125 |
|
|
|
|
0.8% |
>/=20 |
|
|
|
|
0-14 |
420 |
3% |
>/=5 |
23.6 |
126, 127 |
|
|
3-18 |
1212 |
4.1% |
>/=3 |
35.3 |
128, 129 |
(a) Per 100,000 per year in the 0-14 age group
(b) Incidence in Pennslyvania
Table 11.1. The Prevalence of
Islet Cell Antibodies (
New cases of type 1A diabetes presenting in adult life tend to have a longer duration of symptoms before diagnosis and higher C peptide levels remaining at diagnosis compared with those presenting in childhood129, suggesting a slower rate of beta cell destruction. "Slow onset" type 1A diabetes, presenting in late adult life, may be diagnosed initially as type II diabetes. Tuomi and coworkers studied 102 adults who were treated for type II diabetes, of whom one third had low stimulated C peptide levels and two thirds had normal levels130. Of the group with low C peptide, 76% were positive for anti-GAD (similar to the frequency among children with newly diagnosed type 1A diabetes). In comparison, only 12% of this group with normal C peptide was positive for anti-GAD.
In many
studies, it is unknown how long autoantibodies were present in these
individuals before they were discovered.
However, studies by Pilcher and Elliott in New Zealand suggest that ICAs
develop early in childhood and are usually preceded by IAA131. In the studies 666 first-degree relatives,
aged less than 20 years and initially ICA negative (<10 JDF units), were
followed longitudinally. Those under 5
years of age were retested annually, with older subjects retested every 3 to 5
years. Sixteen (2.4%) became
Ziegler and
coworkers in
Both insulin and GAD have been suggested as candidates for the initiating antigen but another (unidentified) autoantigen could be responsible136, 137. Neither IAA nor anti-GAD is universally present among patients with newly diagnosed type 1A diabetes104, 138-140, 140-143. Of note a small subset of children followed from birth in the DAISY study have expressed multiple islet autoantibodies that were lost prior to the onset of diabetes. It is clear that although insulin autoantibodies usually appear first a significant percentage of children followed from birth initially express GAD65 autoantibodies, while IA-2 and ZnT8 autoantibodies are usually the last to develop. With improved assays for insulin autoantibodies81, in the DAISY study insulin autoantibodies are almost always first or present with other autoantibodies and >95% of those progressing to diabetes at some point express insulin autoantibodies (DAISY study, unpublished.)
Several observations provide circumstantial evidence for insulin as an initiating autoantigen. Insulin is the only antigen unique to the beta cell and it is present on the cell surface144, 145. In contrast, GAD is also present in alpha cells146, which are not destroyed15, and insulitis disappears when all insulin-containing cells have been destroyed147. IAAs occur with greater frequency and at higher levels in younger newly diagnosed patients148-150 and the level of IAA correlate with the rate25 of beta cell destruction in prediabetic individuals151. When 29 patients were studied, their IAAs recognized the same epitope152. Williams and coworkers improved the assay for insulin autoantibodies with the development of a microassay153, 154. This assay utilizes protein A or protein A/protein G for autoantibody precipitation and thus avoids the false autoantibody positives for cord or hemolyzed blood associated with the polyethylene glycol based assays. We have modified the Williams assay such that it is performed in 96-well micro titer plates utilizing membrane filtration and direct b-counting in the micro titer plate155. With this assay not only do prediabetic individuals express readily detectable anti-insulin autoantibodies but also NOD mice express high levels of the autoantibody155. Of note administration of a dominant peptide of insulin (insulin B chain peptide B:9-23) induces autoantibodies to insulin, even in Balb/c normal mice that react with intact insulin and are not absorbed by the immunizing peptide156. This autoantibody induction is MHC restricted, suggesting that with the MHC of either Balb/c or NOD mice, T cell activation by an insulin peptide activates already sensitized B-lymphocytes recognizing insulin156. In addition in the NOD mouse model an insulin 1 gene knockout almost completely prevents the development of diabetes, while an insulin 2 gene knockout (the mouse insulin expressed within the thymus) accelerates the development of diabetes157, and NOD mice with both native insulin genes knocked out (with mutated insulin transgene) do not develop diabetes158. Insulin peptide B:9-23 is a dominant NOD target recognized in a specific MHC class II register159, 160
Are there measurable abnormalities that precede development of islet
autoantibodies.
With the existence of many children, both relatives and general population children, followed from birth to the development of diabetes and high throughput, proteomic, metabolomic161, 161-163, messenger RNA assays, as well as improving T cell assays a number of investigators have reported abnormalities that may precede development of islet autoantibodies. This includes reported abnormalities of lipids, metabolites such as glutamate reported from studies in Finland, evaluation of serum effects on messenger RNA expression arrays from Wisconsin, and T cell assays evaluating LADA patients in the state of Washington101, 102, 164 At present with single reports with identification of different metabolites and lack of standardization and utilization of such assays by multiple groups there is not a clear consensus that specific abnormalities precede the development of islet autoantibodies. We personally doubt there will be “non-genetically” determined abnormalities preceding the appearance of islet autoantibodies. Given type 1A diabetes as a T cell mediated autoimmune disorder an analogy would be to search for metabolomic abnormalities preceding anti-polio antibodies induced by polio vaccination.
Hampe and coworkers have reported the presence of anti-idiotypic antibodies that mask the presence of GAD65 autoantibodies in normal controls, with the additional report that such anti-idiotypic antibodies are not present in patients with diabetes, and that individuals progressing to diabetes lose anti-iditiotypic antibodies165. In that the detection of anti-idiotypic antibodies relies upon the use of columns with human monoclonal anti-GAD autoantibodies, We are not convinced as to the actual presence of anti-idiotypic antibodies and this is an area being further studied165 and a recent report correlated changed of anti-idiotypic antibodies with c-peptide changes166, 167.
Are Environmental Factors Important, Either in the Initiation of Beta
Cell Autoimmunity or in Modulating the Process Once It Has Begun?
The diabetes susceptibility genes identified so far are present in a high proportion of the general population, indicating that they are not sufficient to cause the disease. Furthermore, the concordance rate observed in HLA-identical siblings is lower than that in identical twins35, 168 and there are more than 50 loci discovered68, 74, 169. As type 1A diabetes is associated with other autoimmune diseases170, 171, it is known that at least some of the important genes will not be specific to type 1A diabetes but also involved in the susceptibility to other172, 173 autoimmune diseases174 such as celiac disease175 and rheumatoid arthritis176.
Despite the importance of genetic factors, the concordance rate is less than 100% in identical twins148-150, 177 and the rapidly increasing incidence of Type 1 diabetes in Western societies178 implies a critical role for non-genetic random re-arrangement of T cell receptor or immunoglobulin genes during the differentiation of T and B lymphocytes179, somatic mutation, and or environmental factors. Epidemiological studies with validated case ascertainment support a role for environmental factors. A rising incidence of type 1A diabetes over time, too rapid to be explained by an increase in the frequency of diabetogenic genes in the population, has been reported in multiple countries180-189. In addition, increased rates have been observed in migrant populations compared with their countries of origin190, though the change in migrant populations is relatively small191.
An
environmental factor could be involved either by initiating autoimmunity or by
altering its progression, once established.
There is a seasonal variation in the onset of type 1A diabetes123, 126, 180, 181, 192,
with more cases in the winter months, associated with viral infections. It is likely that such viral infections alter
the process late in its course by increasing insulin requirements, thereby
advancing the time of diabetes onset. In
humans, the best evidence for the involvement of the specific viral agent in
the initiation of autoimmunity comes from the observation that patients with
congenital rubella have an increased prevalence of type 1A diabetes193, 194,
as well as other autoimmune diseases195. Of note anti-islet autoantibodies are
relatively uncommon196.
Coxsackievirus B infection (enterovirus197, 198)
has also been suggested as an environmental trigger199, although a large Swedish
study found no significant difference in the levels of IgM antibodies against
Coxsackievirus B types 1-5 between newly diagnosed diabetic children and
population controls200. There is likely to be a long lag time between
a triggering infection and the onset of diabetes (perhaps short time between
infection and anti-islet autoantibody induction[weeks])but IgG levels were not
measured in this study. It has been
suggested that an infectious agent might activate autoimmunity by molecular
mimicry, whereby the immune response to a viral antigen, such as the P2-C
protein of Coxsackievirus B4201 or the rubella capsid202, might cross-react with a
beta cell antigen. Some evidence against
a significant role for an infectious agent comes from
At present
studies of potential environmental factors triggering the development of
anti-islet autoantibodies in children followed from birth have not led to the
identification of major precipitating factors, and contradictory results are a
problem210. Studies from
In oral communications Dotta and coworkers have reported the finding of enterovirus immunoreactivity within islets that have died with new onset diabetes. The number of pancreases analyzed is small (approximately 6) but ½ of patients had enteroviral antigen within islets,. Apparently an enterovirus sequenced was a laboratory related sequence. The presence of viruses in a subset of pancreases of patients with type 1 diabetes is an active area of investigation.
In addition, in animal models of type 1 diabetes, exposure to infectious agents may suppress rather than trigger autoimmunity. Diabetes-prone BB rat housed in "viral antibody free" conditions have a higher frequency of diabetes than those housed in less stringent "specific pathogen free" conditions222. In contrast NOD mice with a specific mutation blocking toll receptor signaling have autoimmune diabetes dependent upon specific gut flora223. Specific infection with mouse hepatitis virus is associated with reduced incidence of diabetes in the NOD mouse224, and infection with lymphocytic choriomeningitis virus is associated with reduced incidence in both the NOD mouse and BB rat225, 226. However, in at least one instance infection is implicated as a trigger. Kilham's rat virus, a parvovirus, has been shown to trigger beta cell autoimmunity and diabetes in the diabetes-resistant BB rat227. It appears that the Kilham virus acts similar to poly-IC rather than by infecting islet cells228. Other reports of viruses triggering diabetes in animals may involve direct viral damage of beta cells without an autoimmune mechanism229 or bystander stimulation230, 231.
In humans, associations with infant feeding have been reported. Several population-based case-control studies found a protective effect associated with breast-feeding232-234 and an increased risk associated with the early introduction of supplemental feeding232, 233, 235. A Danish study, which eliminated several possible sources of bias by using the records of postnatal health visitors, found no evidence to support an associated with the total duration of breast feeding236, but did not examine the timing of introduction of supplemental feeding. Both BabyDiab and DAISY found no evidence for infant bovine milk ingestion213 but a pilot study of the elimination of bovine milk from infant formula was associated with a reduction of development of cytoplasmic ICA, with little reduction of GAD65 autoantibodies212.
The reported associations with infant feeding are weak (odds ratios ~1.5)237, but this may be consistent with the effect of a common exposure acting only on genetically susceptible individuals. Several mechanisms could explain the associations with infant feeding. Breast-feeding could protect the infant from an infection. The increased caloric intake and weight gain associated with artificial feeding might cause increased insulin secretion and presentation of beta cell autoantigens238. Karjalainen and coworkers have suggested that intact cow's milk peptides might cross the immature gut, initiating an immune response to cross-reacts with a beta cell surface antigen(146). Higher levels of antibodies against several components of cow's milk have been reported in children with newly diagnosed type 1A diabetes than in controls239-243. T cell proliferation in response to the ABBOS peptide was higher in newly diagnosed patients than in controls244. These results could not be reproduced by Atkinson and coworkers(153).They found no difference between newly diagnosed patients and controls for either anti-BSA antibodies or T cell proliferation to ABBOS. In animal studies, the addition of cow's milk to the diet of the BB rat at the time of weaning increases the incidence of diabetes245, 246, but there are conflicting reports on the effect of cow's milk in the diet of the NOD247, 248. Of interest there is insulin in both human milk and bovine milk and a reported induction of anti-bovine insulin antibodies with bovine milk ingestion249 as well as an initial clinical trial of removing insulin from milk 250.
Ziegler and coworkers and Norris and coworkers (figure 11.10) have reported in JAMA an association with the induction of anti-islet autoantibodies with the introduction of cereals/gluten prior to three months of age251, 252. In the report by Norris and coworkers, there was a biphasic association with autoantibodies, namely increased risk with introduction before 3 months and after 6 months of age. Decreased omega-3-fatty acid consumption has been reported to be associated with increased risk in the DAISY study253, 254. The National Institutes of Health have established a major international consortium termed TEDDY (The Environmental Determinants of Type 1 Diabetes in the Young) that will follow infants from birth for the development of anti-islet autoimmunity and diabetes75. No association with development of islet autoantibodies with Vitamin D intake was found in the DAISY study255.
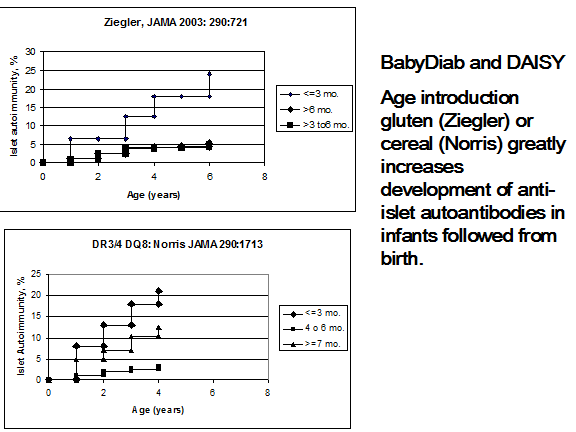
Figure 11.10: Life table analysis of expression of anti-islet autoantibodies for infants followed from birth relative to the age of introduction of cereal/gluten.
Once Autoimmunity Is Initiated, Are Beta Cells Destroyed in a
Progressive, Linear Fashion or Are There Spontaneous Remissions?
The long-term follow-up of high-titer ICA-positive relatives in the Joslin family study with serial IVGTTs suggested a model in which beta cell destruction is a progressive, linear process256, 256-258 . The rate of beta cell destruction may vary widely from one individual to another. It may be so slow in some individuals that such overt diabetes does not develop during the person's lifetime. Such individuals might appear to have decreased but stable beta cell function when assessed with serial IVGTTs over many years. A correlation between serial FPIR levels and the number of years to diabetes in ICA-positive relatives259, and the addition of IAA as a risk marker260, led to the development of a "dual parameter" linear regression model151. In this model the time remaining before the onset of diabetes is predicted by a combination of the FPIR (reflecting the remaining beta cell mass) and the IAA level (reflecting the rate of loss of beta cells). Recent analysis of children followed from birth indicates age at appearance of islet autoantibodies and mean levels of insulin autoantibodies predict approximate age of diabetes onset25.
Other studies,
including relatives with lower titer
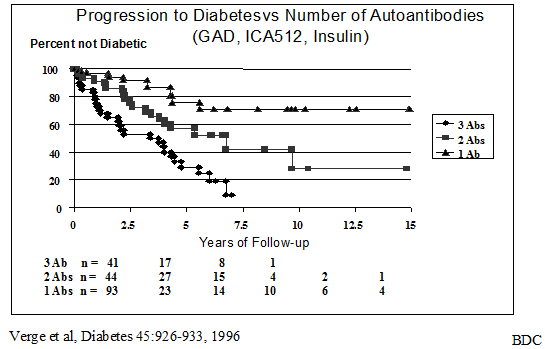
Figure 11.11 Progression of autoantibody positive first degree relatives to diabetes, subdivided by the number of biochemical autoantibodies expressed.
Low-titer ICAs carry a reduced risk of type 1A diabetes, compared with high titer27, 28, and it may be that fluctuating levels do not signify a disease process, especially unaccompanied by other autoantibodies. Non-persistent CAs may represent a nonspecific immunological response associated with infection, as Helmke and coworkers reported the transient appearance of ICAs in normal schoolchildren during mumps infection268. The development of more reproducible assays for antibodies against biochemically defined autoantigens may improve the assessment of diabetes risk. For relatives who express more than one autoantibody, levels of IAA59, anti-GAD, and anti-ICA512 can be remarkably persistent over time11, 271, 276.
Carel and coworkers reported that ICA-negative siblings of diabetic children have significantly lower mean FPIR than controls277. They suggested that autoimmune beta cell destruction may have been initiated in some of these siblings, but spontaneously remitted. However, other autoantibodies may have been present in some of the ICA-negative siblings in this study. At most, only 94% of children with newly diagnosed type 1A diabetes are ICA positive, using a very sensitive assay278 but the addition of other autoantibodies may reduce the autoantibody-negative proportion. In addition, although the diet was standardized for the 3 days prior to the IVGTT, there may be differences over the longer term in the diets of children with and without a diabetic sibling that could have affected the IVGTT results.
Experiments with streptozotocin-treated baboons indicate that beta cell function (assessed by IVGTT) correlates with beta cell mass279. In humans, however, the IVGTT has several limitations. The results of beta cell function tests may vary with insulin sensitivity according to age277, 280, physical fitness281, body mass index280, 282, pubertal status282, 282, 283, 283, 283, and physical or psychological stress. Different protocols for the IVGTT give different results284 but a standard protocol has been developed to allow meta-analyses285.
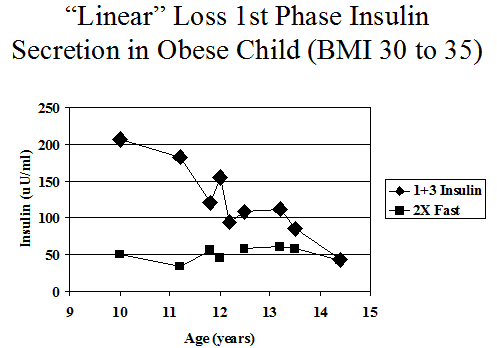
Figure 11.12 Intravenous glucose tolerance insulin levels of antibody positive markedly obese female progressing to diabetes with fasting insulin typically above 25 (illustration 2 times fasting insulin) and sum 1+3 minute declining progressively, until there is no first phase secretion at onset of diabetes.
The interpretation of serial FPIR data is complicated by within-subject variability in the measurement. Figures 11.12 and11.13 illustrates the course of the FPIR over time for two ICA-positive relatives. One has a clearly linear loss of secretion (Fig. 11.12). In contrast, the other relative (Fig. 11.13) has what appears to be a stepwise loss of secretion, with remissions and periods of recovery. However, this second pattern could result
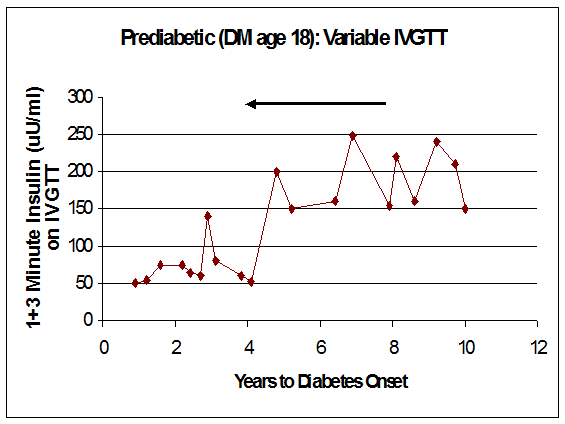
Figure 11.13 Marked variation in loss of first phase insulin secretion on intravenous glucose tolerance testing during progressing to diabetes (years prior to diabetes from right to left).
from wide within-subject measurement variation, superimposed on an underlying loss of secretion. In the large DPT-1 study loss of first phase insulin secretion was strongly associated with diabetes risk57 as it was in the combined ICARUS dataset58 and with large datasets considerable variability exists in the patterns of loss of first phase insulin secretion prior to diabetes. A significant percentage of prediabetic individuals have retained first phase secretion within one year of diabetes, with many of these appearing to have high fasting insulin and potentially severe insulin resistance. Recent data in the youngest children expressing anti-islet autoantibodies indicates that at the time of detection of autoantibodies many of those children already have severely depressed FPIR, though a higher risk for diabetes is also in that study associated with an average earlier onset of diabetes56 (Figure 11.14).
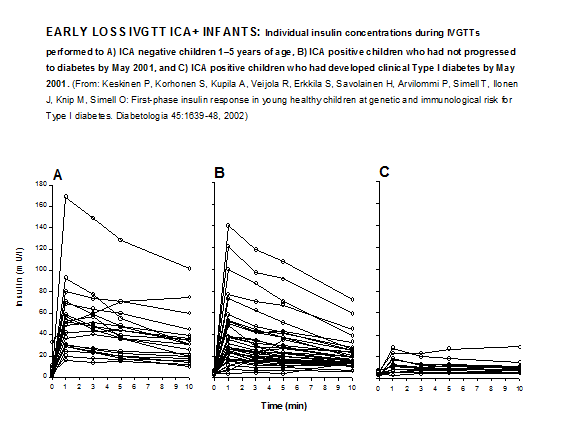 Figure 11.14 Early loss IVGTT response in
Figure 11.14 Early loss IVGTT response in
Several centers have performed duplicate IVGTTs, spaced 1 or more weeks apart, on healthy volunteers. The median within-subject coefficients of variation (CV) reported for FPIR varied from 4% to 36%280, 286, 287 but the CV was as high as 56% for some subjects. There are conflicting reports about whether calculating the area under the curve for insulin release improves reproducibility, compared with a sum of the 1- and 3-minute insulin levels280, 286, 287. McCulloch and coworkers studied 18 normal post pubertal subjects on two occasions separated by an average of 1 year261. They used an IVGTT protocol with frequent sampling and a bolus of tolbutamide at 20 minutes, allowing the calculation of the insulin sensitivity index. The within-subject CV for the incremental area under the insulin curve (above fasting levels) from 0 to 10 minutes was 11.3%. Adjusting for the insulin sensitivity index resulted in a similar CV (10.5%). However, it is likely that adjusting for insulin sensitivity may improve reproducibility in pubertal subjects and those followed over a longer period. The subjects also underwent arginine stimulation tests (both at physiological plasma glucose concentration and hyperglycemic clamp). The reproducibility of the response to arginine was similar to that of the response to glucose, indicating little advantage gained by the use of arginine.
Rayman and coworkers achieved highly reproducible results using retrograde venous cannulation and arterialization of the hand from which the samples are drawn286, but recent studies found no improvement with these techniques288, 289. Other maneuvers that have been evaluated to improve reproducibility include placement of the intravenous line 1 hour before the test to minimize the effects of stress hormones. Allen and coworkers tested the hypothesis that catecholamine release, associated with anxiety about the test, may inhibit insulin release but found no correlation between FPIR and plasma catecholamine levels or standardized anxiety scores280. However, a significant "first test effect" was observed. Among 11 normal subjects who underwent two tests, 9 had a higher FPIR on the second test280. One subject who fainted during the first test had a very low FPIR (2 mU/L) that became normal on a subsequent occasion.
Though loss of first phase insulin secretion is a sensitive marker of beta cell abnormalities and aids in the prediction of diabetes it is a labor intensive test. At present in the DAISY study we follow autoantibody positive individuals with measurement of HbA1c with fingerstick blood sampling. For the great majority of children progressing to diabetes HbA1c begins to rise one to several years prior to the onset of diabetes in the normal range290. When HbA1c approaches the upper limit or exceeds the upper limit of normal we perform a formal oral glucose tolerance test to confirm the diagnosis of diabetes. In addition fasting glucose, glucose at 120 minutes on oral glucose tolerance testing, and C-peptide on oral glucose tolerance testing can all be used to stage progression to diabetes of islet autoantibody positive individuals24673}. HbA1c >6.5% has an approximate 25% sensitivity to identify early diabetes in prospective studies with high specificity291.
The FPIR is a measure of beta cell function, but a measure of beta cell mass might be more useful. In the future, new ways of assessing the beta cells may be developed, including high-performance glycated hemoglobin assays and imaging with nuclear magnetic resonance (MRI) or isotopic scans. Leech and coworkers measured the HbA1c by high-performance liquid chromatography and healthy teenagers who were also tested for ICA292. The mean HbA1c was slightly, but significantly, higher in the ICA-positive subjects, suggesting that elevation of the HbA1c within the normal range may be an early marker of loss of beta cell function. Signore and coworkers reported preliminary data suggesting that scans following the injection of293 I-labeled interleukin-2 (IL-2) may be useful in the detection of beta cell autoimmunity294. A significant accumulation of tracer was detected in the pancreatic region of four newly diagnosed and two prediabetic subjects, compared with four controls. MRI has been used to detect pancreatic graft rejection295-297. With refinements in MRI techniques it may be possible in the future to detect insulitis or to approximate beta cell mass298, 299. In the NOD mouse model with its extensive insulitis several methods for detecting insulitis using iron particles have been developed. One apparently measures vascular leakage and the other specific binding of autoantigenic T cells to their cognate receptor. Studies to apply such techniques to man are underway, but the amount of insulitis given current analysis of pancreas from patients of the nPOD study indicate that insulitis can be be minimal300. In addition several groups are attempting to develop technology for non-invasive imaging of beta cell mass with at present controversial results301.
There is little information on changes in islet histology before the onset of overt diabetes in humans. However, Sutherland and coworkers studied patients with longstanding type 1A diabetes who received pancreatic transplants from their non-diabetic identical twins147. Of these, three received either delayed immunosuppression or no immunosuppression after the transplants and all three became insulin dependent again within 5 to 12 weeks. Serial biopsies of the graft revealed T cell infiltration with selective beta cell destruction, unlike graft rejection. Rather, these histological findings were consistent with reactivation of beta cell autoimmunity. Of note, the insulitis disappeared after the insulin-containing cells were destroyed.
Foulis and
coworkers studied the pancreatic histology of 119 patients with type 1 diabetes
who had died of ketoacidosis, of whom 60 had died within 1 year of diagnosis15. More recent studies from
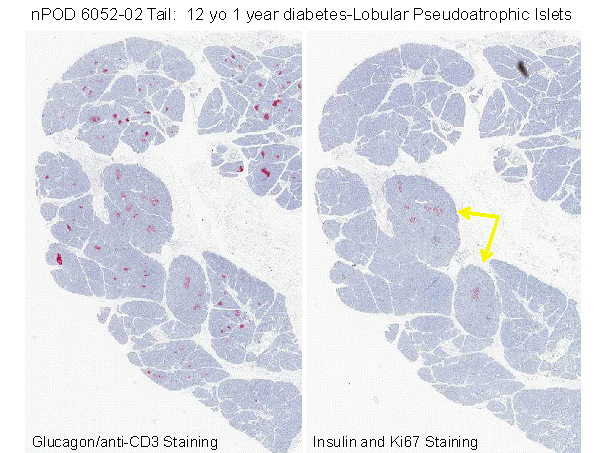
Figure 11.15 Pancreatic section from nPOD website showing marked atrophy of acinar pancreas in area where all beta cells within islets are destroyed (right panel) versus lack of atrophy on left of section where 100% of islets have extant beta cells. Inset illustrates vitiligo on legs of patients with similar lobular loss of target cells, in this case melanocytes. Suggest that Type 1A diabetes develops as vitiligo of the pancreas with immune mediated lobular killing of beta cells.
The diagnosis of diabetes is to some extent an arbitrary point in the natural history of the autoimmune disease. The timing of overt diabetes may be influenced by factors that increase insulin requirements, such as infections, obesity and puberty. Recent studies indicate that insulin secretion in normal individuals and individuals at risk for type 1 diabetes who do not progress to diabetes usually increases with age10. This is likely related to increasing insulin resistance with age, and thus when an individual remains with stable insulin secretion (e.g. C-peptide secretion on mixed meal tolerance test) or with decreasing insulin secretion, marked functional abnormalities of beta cell function are present. Wilkins and coworkers have hypothesized (Accelerator Hypothesis)309 that type 1 and type 2 diabetes are essentially the same disorder with three factors “accelerating” loss of beta cells in type 1 diabetes (high rate of beta cell apoptosis; insulin resistance; beta cell autoimmunity)310. Given the increasing understanding of the genetics of type 1A diabetes and ability to predict the disease with immunologic assays, both of which are lacking in type 2 diabetes, we believe it is more likely that insulin resistance may impact development of type 1 diabetes simply by influencing the timing of hyperglycemia in the presence of limited insulin secretion. Such insulin resistance may relate to reports relating BMI and higher energy intake prior to type 1 diabetes311, 312. Consistent with this, insulin resistance in analysis of the ENDIT study influenced progression only in those with low insulin secretion313. Experiments with streptozotocin-treated baboons suggest that the FPIR may be undetectable even when about 20%-50% of beta cell mass remains279, although pancreatic insulin content was close to zero at this point. Autoimmune beta cell destruction continues after the diagnosis of diabetes. A temporary remission from insulin dependency may occur in up to 27% of patients, soon after diagnosis314, and is attributed to the beta cell rest caused by insulin treatment and slowing of loss of C-peptide is associated with intensive insulin therapy in the DCCT trial10. Younger age at onset, male sex, high-titer ICA, severe ketoacidosis at diagnosis, and a short duration of symptoms prior to diagnosis are associated with a more rapid loss of C peptide secretion315. In one study a more rapid loss was seen in patients heterozygous for DR3 and DR4316, although another study came to the opposite conclusion314, and another found no significant DR associations315. It is likely that insulin resistant individuals present earlier with overt diabetes for any given loss of beta cell mass and increasing fasting glucose prior to onset of diabetes has been reported, a trend toward increasing fasting insulin is also found, and most important impaired glucose tolerance on oral glucose tolerance testing10 frequently precedes overt diabetes 259.
Studies by
Conclusions
We propose a model in which immunological abnormalities appear early in life in many genetically susceptible individuals but in which anti-islet autoimmunity can remain discordant for years for both children and adults for genetically identical individuals. The presence of autoimmunity is most easily marked by the appearance high affinity anti-islet autoantibodies. Anti-insulin autoantibodies when first present in children followed from birth are already of high affinity. Environmental triggers activating innate immunity and T cells may rapidly (weeks) activate autoimmunity206, 207, 221. Alternatively autoimmunity may be aborted by environmental factors (e.g., various infections). In a subset of individuals, these abnormalities may be transient and are not associated with beta cell destruction, though once multiple autoantibodies are present on more than one occasion almost all individuals progress to diabetes, even though the specific autoantibodies positive varies over years. Once beta cell destruction is initiated, marked by the appearance of multiple immunological abnormalities and later by measurable loss of FPIR and progressive loss of glucose tolerance. Anti-islet autoantibodies can be present for decades without loss of insulin secretion in some individuals, but over time most individuals expressing multiple biochemical anti-islet autoantibodies progress to overt diabetes. Genetic and environmental factors may modulate the process, affecting the rate of beta cell destruction. There is not a clear pattern in the variation with some autoantibodies rising in level while others are falling with a time course often measured in years. Levels of only insulin autoantibodies correlate with rate of progression to diabetes25 and since these levels can vary over time, the rate of destruction of beta cells likely varies. The loss of beta cells may be so slow in some individuals that overt diabetes does not occur during the person's lifetime. The presence of more than one of these autoantibodies (GAD65, ICA512, ZnT8, or insulin), combined with FPIR or glucose tolerance measurements and typing for HLA-DQ alleles, allows the identification of a subset of relatives with sufficiently high-risk for type 1A diabetes to carry out preventive trials.
The hypothesis that type 1A diabetes developed in a chronic and predictable manner forms the basis for prevention trials. It is equally important to identify those autoantibody-positive relatives who are unlikely to progress to diabetes (e.g. great majority of those with a single autoantibody). Knowledge about the natural history of the prediabetic period may also facilitate the diagnosis of type 1A diabetes. Such information becomes clinically relevant in genetic counseling, the diagnosis of diabetes in adults (type 1A versus type II), and in children with an unusual clinical course.
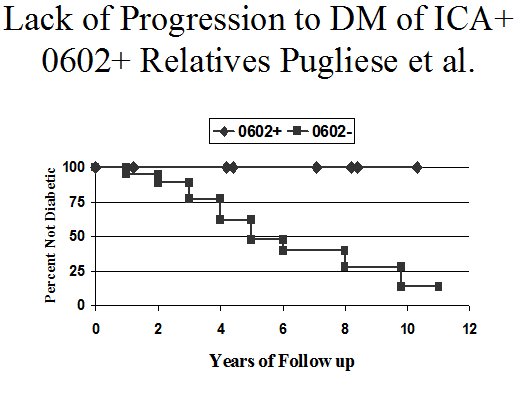
Figure 11.16 Lack of progression to diabetes of
Evaluation of each of the above circumstances will usually include genetic, immunological, and metabolic information. The presence of the HLA allele DQA1*0102/DQB1*0602 is so rare (approximately 1%) among children with type 1A diabetes318, 319 that an alternative disorder (non Type 1A) should be considered as the cause of diabetes if it’s haplotype is present(Figure 11.16). Lack of all biochemical autoantibodies at onset in children should also stimulate search for unusual forms of diabetes320. Other diseases that can present as insulin-requiring diabetes in childhood include Kir6.2 mutations (sulfonylurea therapy may suffice) for “neonatal” diabetes321 Wolfram's syndrome (DIDMOAD-diabetes insipidus, diabetes mellitus, optic atrophy, and nerve deafness)322, mitochondrial mutations (often associated with nerve deafness323), and MODY324 (maturity onset diabetes of youth, that may be caused by mutations in the glucokinase and HLA genes)(204). The DQB1*0602 protective haplotype is likely to be present in these disorders at the normal population frequency of approximately 20%.
In adults, the
presence of specific autoantibodies in a diabetic individual is probably the
best available evidence of type 1A diabetes.
At an epidemiological level, the diagnosis of type 1A diabetes by
presence of ketoacidosis, insulin requirement, and young age of onset is
useful, but for an individual may be of little value91, 325. In the past, determination of
Of 63 children with transient hyperglycemia we have evaluated, most (89%) remain non-diabetic327. All of the few with ICAs or IAAs became permanently diabetic within 18 months. It is likely that the measurement of antibodies against multiple biochemically defined antigens will also improve diagnosis of this group. The IVGTT is the diagnostic test choice in this group. Among children with resolved transient hyperglycemia, none of those with normal FPIR (greater than the 5th percentile) has progressed to diabetes, though with increasing insulin resistance in the population there are bound to be exceptions, and we usually subtract 2*fasting insulin from the 1+3 minute insulin following intravenous glucose as a measure of FPIR. Of those with low FPIR, a subset normalizes upon repeat testing, but those remaining abnormal have all become overtly diabetic within 1 year.
An individual contemplating the donation of a kidney to a diabetic relative should have an OGTT to rule out the presence of overt diabetes, an IVGTT by ICARUS criteria, and determination of autoantibodies with assays that have high specificity and sensitivity (e.g., the combination of anti-GAD, anti-insulin, anti-ZnT8 and anti-ICA512). Most of these assays are available now in commercial laboratories.
Finally, the
most important reason for detecting individuals at risk for type 1A diabetes is
the potential for preventive therapy. It
is likely that with more detailed analysis and improvements in the assays
available, the prediction of type 1A diabetes will improve further. Sosenko and coworkers have developed a series
of combined risk scores for autoantibody positive individuals53, 54. It will be very important to begin such
prediction in the general population, as the majority of new cases have no
affected relative. The large ENDIT trial demonstrated the feasibility of
international trials based upon screening for cytoplasmic islet cell
autoantibodies, though nicotinamide did not influence progression to diabetes328.
The large DPT-1 trial (parenteral and oral insulin therapy trials) is now
complete and this study with more than 90,000 first degree relatives screened
for islet autoantibodies provides a wealth of information. Parenteral and oral insulin therapy did not
slow progression to diabetes though a subset of individuals entering the oral
trial with high levels of insulin autoantibodies may have had a slowing of
progression to diabetes and TrialNet is planning a study to confirm or refute
this observation329, 329. Initial analysis of more than 70,000
relatives tested for GAD65 and ICA512 autoantibodies confirms the predominant
influence of presence of multiple autoantibodies for prediction43. One half of cytoplasmic ICA positive
relatives did not express any of the biochemical autoantibodies, and just as
important the cytoplasmic ICA assay failed to detect GAD65 or ICA512
autoantibodies in approximately 2% of the relatives43. The presence of biochemical anti-islet
autoantibodies at the initial screening was associated with high risk for
eventual eligibility for the DPT trial (e.g. low first phase insulin
secretion). The presence of a single biochemical
autoantibody and abnormal glucose tolerance will likely identify an additional
high-risk group. Presence of
A very large
NIH effort is underway to prevent type 1A diabetes, including TrialNet, the
Immune Tolerance Network,
Reference List
1.
Eisenbarth GS. Prevention of Type 1A
Diabetes. Endocr Pract 2012;1-17.
2.
Skyler JS. Immune intervention for type
1 diabetes mellitus. Int J Clin Pract Suppl 2011;(170):61-70.
3.
Todd JA, Knip M, Mathieu C. Strategies
for the prevention of autoimmune type 1 diabetes. Diabet Med
2011;28(10):1141-1143.
4.
Flanders G, Graves P, Rewers M.
Prevention of type 1 diabetes from laboratory to public health. [Review] [91
refs]. Autoimmunity 1999;29(3):235-246.
5.
Ziegler AG, Nepom GT. Prediction and
pathogenesis in type 1 diabetes. Immunity 2010;32(4):468-478.
6.
Noble JA, Valdes AM. Genetics of the HLA
region in the prediction of type 1 diabetes. Curr Diab Rep 2011;11(6):533-542.
7.
Yu L, Boulware DC, Beam CA et al. Zinc
Transporter-8 Autoantibodies Improve Prediction of Type 1 Diabetes in Relatives
Positive for the Standard Biochemical Autoantibodies. Diab care 2012.
8.
Gianani R, Eisenbarth GS. The stages of
type 1A diabetes: 2005. Immunol Rev 2005;204:232-249.
9.
Keenan HA, Sun JK, Levine J et al.
Residual insulin production and pancreatic ss-cell turnover after 50 years of
diabetes: Joslin Medalist Study. diab 2010;59(11):2846-2853.
10.
Sherry NA, Tsai EB, Herold KC. Natural
History of {beta}-Cell Function in Type 1 Diabetes. Diabetes 2005;54 Suppl
2:S32-9.:S32-S39.
11.
Achenbach P, Bonifacio E, Koczwara K,
Ziegler AG. Natural history of type 1 diabetes. Diabetes 2005;54 Suppl
2:S25-31.:S25-S31.
12.
Haller MJ, Atkinson MA, Schatz D. Type 1
diabetes mellitus: etiology, presentation, and management. Pediatr Clin North
Am 2005;52(6):1553-1578.
13.
Eisenbarth GS. Italian Society of
Diabetology Mentor Award. The stages of Type 1A diabetes: retrospective and
prospective. Diabetes Nutr Metab 2004;17(6):374-385.
14.
Eisenbarth GS. Prediction of type 1
diabetes: the natural history of the prediabetic period. Adv Exp Med Biol
2004;552:268-90.:268-290.
15.
Foulis AK, Liddle CN, Farquharson MA,
Richmond JA, Weir RS. The histopathology of the pancreas in type I diabetes
(insulin dependent) mellitus: a 25-year review of deaths in patients under 20
years of age in the United Kingdom. diabetol 1986;29(5):267-274.
16.
Foulis AK, Farquharson MA, Hardman R.
Aberrant expression of class II major histocompatibility complex molecules by b cells and hyperexpression of class
I major histocompatibility complex molecules by insulin containing islets in
type 1 (insulin dependent) diabetes mellitus. diabetol 1987;30(5):333-343.
17.
Gepts W. Pathologic anatomy of the
pancreas in juvenile diabetes mellitus. diab 1965;14(10):619-633.
18.
Komulainen J, Knip M, Lounamaa R et al.
Poor beta-cell function after the clinical manifestation of type 1 diabetes in
children initially positive for islet cell specific autoantibodies. The
Childhood Diabetes in Finland Study Group. Diabet Med 1997;14(7):532-537.
19.
Pozzilli P, Pitocco D, Visalli N et al.
No effect of oral insulin on residual beta-cell function in recent-onset type I
diabetes (the IMDIAB VII). IMDIAB Group [In Process Citation]. diabetol
2000;43(8):1000-1004.
20.
Chaillous L, Lefevre H, Thivolet C et
al. Oral insulin administration and residual beta-cell function in recent-onset
type 1 diabetes: a multicentre randomised controlled trial. Lancet
2000;356(9229):545-549.
21.
Klinke DJ. Age-corrected beta cell mass
following onset of type 1 diabetes mellitus correlates with plasma C-peptide in
humans. PLoS ONE 2011;6(11):e26873.
22.
Greenbaum CJ, Beam CA, Boulware D et al.
Fall in C-peptide During First 2 Years From Diagnosis: Evidence of at Least Two
Distinct Phases From Composite TrialNet Data. diab 2012.
23.
Meier JJ, Bhushan A, Butler AE, Rizza
RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing
type 1 diabetes: indirect evidence for islet regeneration? Diabetologia
2005;48(11):2221-2228.
24.
Ikegami H, Ogihara T. Genetics of insulin-dependent
diabetes mellitus. Endocr J 1996;43:605-613.
25.
Steck AK, Johnson K, Barriga KJ et al.
Age of islet autoantibody appearance and mean levels of insulin, but not GAD or
IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes
autoimmunity study in the young. Diab care 2011;34(6):1397-1399.
26.
Parikka V, Nanto-Salonen K, Saarinen M
et al. Early seroconversion and rapidly increasing autoantibody concentrations
predict prepubertal manifestation of type 1 diabetes in children at genetic
risk. diabetol 2012.
27.
Riley WJ, Maclaren NK, Krischer J et al.
A prospective study of the development of diabetes in relatives of patients
with insulin-dependent diabetes. N Engl J Med 1990;323(17):1167-1172.
28.
Bonifacio E, Bingley PJ, Shattock M et
al. Quantification of islet-cell antibodies and prediction of insulin-dependent
diabetes. Lancet 1990;335(8682):147-149.
29.
Verge CF, Gianani R, Kawasaki E et al.
Number of autoantibodies (against insulin, GAD or ICA512/IA2) rather than particular
autoantibody specificities determines risk of type I diabetes. J Autoimmun
1996;9(3):379-383.
30.
Verge CF, Stenger D, Bonifacio E et al.
Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin
autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes:
Combinatorial Islet Autoantibody Workshop. diab 1998;47(12):1857-1866.
31.
Strebelow M, Schlosser M, Ziegler B,
Rjasanowski I, Ziegler M. Karlsburg Type I diabetes risk study of a general
population: frequencies and interactions of the four major Type I diabetes-
associated autoantibodies studied in 9419 schoolchildren. diabetol
1999;42(6):661-670.
32.
Christie MR, Roll U, Payton MA, Hatfield
EC, Ziegler AG. Validity of screening for individuals at risk for type I
diabetes by combined analysis of antibodies to recombinant proteins. Diab care
1997;20(6):965-970.
33.
Vardi P, Crisa L, Jackson RA et al.
Predictive value of intravenous glucose tolerance test insulin secretion less
than or greater than the first percentile in islet cell antibody positive
relatives of type I (insulin-dependent) diabetic patients. diabetol
1991;34:93-102.
34.
Sosenko JM, Palmer JP, Rafkin-Mervis L
et al. Glucose and C-peptide Changes in the Peri-Onset Period of Type 1
Diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 2008.
35.
Deschamps I, Khalil I. The role of DQ
alpha--beta heterodimers in genetic susceptibility to insulin-dependent
diabetes. Diabetes Metab Rev 1993;9:71-92.
36.
Verge CF, Gianani R, Kawasaki E et al.
Prediction of type I diabetes in first-degree relatives using a combination of
insulin, GAD, and ICA512bdc/IA-2 autoantibodies. diab 1996;45(7):926-933.
37.
Bingley PJ, Christie MR, Bonifacio E et
al. Combined analysis of autoantibodies improves prediction of IDDM in islet
cell antibody-positive relatives. diab 1994;43(11):1304-1310.
38.
Naserke HE, Ziegler AG, Lampasona V,
Bonifacio E. Early development and spreading of autoantibodies to epitopes of
IA-2 and their association with progression to type 1 diabetes. J Immunol
1998;161(12):6963-6969.
39.
Kimpimaki T, Kulmala P, Savola K et al.
Disease-associated autoantibodies as surrogate markers of type 1 diabetes in
young children at increased genetic risk. Childhood Diabetes in Finland Study
Group. J Clin Endocrinol Metab 2000 Mar ;85 (3 ):1126 -32 2000;85(3):1126-1132.
40.
Pietropaolo M, Peakman M, Pietropaolo SL
et al. Combined analysis of GAD65 and ICA512(IA-2) autoantibodies in organ and
non-organ-specific autoimmune diseases confers high specificity for
insulin-dependent diabetes mellitus. J Autoimmun 1998;11:1-10.
41.
Wenzlau JM, Juhl K, Yu L et al. The
cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1
diabetes. Proc Natl Acad Sci U S A 2007;104(43):17040-17045.
42.
NIH Consensus Development Conference on
Celiac Disease. NIH Consens State Sci Statements 2004;21(1):1-23.
43.
Yu L, Cuthbertson DD, Maclaren N et al.
Expression of GAD65 and Islet Cell Antibody (ICA512) autoantibodies amongst
cytoplasmic ICA+ relatives is associated with eligibility for Diabetes
Prevention Trial Type-1. diab 2001;50:1735-1740.
44.
Williams AJ, Bingley PJ, Moore WP, Gale
EA. Islet autoantibodies, nationality and gender: a multinational screening
study in first-degree relatives of patients with Type I diabetes. diabetol
2002;45(2):217-223.
45.
Krischer JP, Schatz D, Riley WJ et al.
Insulin and islet cell autoantibodies as time-dependent covariates in the
development of insulin-dependent diabetes: a prospective study in relatives. J Clin
Endocrinol Metab 1993;77(3):743-749.
46.
Maclaren N, Lan M, Coutant R et al. Only
multiple autoantibodies to islet cells (ICA), insulin, GAD65, IA-2 and IA-2beta
predict immune-mediated (Type 1) diabetes in relatives. J Autoimmun
1999;12(4):279-287.
47.
Srikanta S, Ganda OP, Gleason RE,
Jackson RA, Soeldner JS, Eisenbarth GS. Pre-type I diabetes. Linear loss of
beta cell response to intravenous glucose. diab 1984;33(8):717-720.
48.
Srikanta S, Ganda OP, Jackson RA et al.
Pre-type I (insulin dependent) diabetes: common endocrinological course despite
immunological and immunogenetic heterogeneity. diabetol 1984;27(Suppl):146-148.
49.
Jackson RA, Vardi P, Herskowitz RD,
Soeldner JS, Eisenbarth GS. Dual parameter linear model for prediction of onset
of type I diabetes in islet cell antibody positive relatives. Clin Res 36,
585A. 1988.
Ref Type: Abstract
50.
Vardi P, Dib S, Herskowitz RD, Wolfsdorf
JI, Soeldner JS, Eisenbarth GS. Multiparameter prediction of months to overt
diabetes in ICA positive first degree relatives. Diabetes 37(Suppl 21A). 1988.
Ref Type: Abstract
51.
Sosenko JM, Skyler JS, Krischer JP et
al. Glucose excursions between states of glycemia with progression to type 1
diabetes in the diabetes prevention trial-type 1 (DPT-1). diab
2010;59(10):2386-2389.
52.
Sosenko JM, Palmer JP, Rafkin LE et al.
Trends of earlier and later responses of C-peptide to oral glucose challenges
with progression to type 1 diabetes in diabetes prevention trial-type 1
participants. Diab care 2010;33(3):620-625.
53.
Sosenko JM, Skyler JS, Mahon J et al.
The Application of the Diabetes Prevention Trial-Type 1 Risk Score for
Identifying a Preclinical State of Type 1 Diabetes. Diab care 2012.
54.
Sosenko JM, Skyler JS, Mahon J et al.
Validation of the Diabetes Prevention Trial-Type 1 Risk Score in the TrialNet
Natural History Study. Diab care 2011;34(8):1785-1787.
55.
Eisenbarth GS, Verge CF, Allen H, Rewers
MJ. The design of trials for the prevention of IDDM. diab 1993;42:941-947.
56.
Keskinen P, Korhonen S, Kupila A et al.
First-phase insulin response in young healthy children at genetic and
immunological risk for Type I diabetes. diabetol 2002;45(12):1639-1648.
57.
Chase HP, Dolan LM, Krischer JP et al.
First phase insulin release during the intravenous glucose tolerance test as a
risk factor for type 1 diabetes. J Pediatr 2001;138(2):244-249.
58.
Bingley PJftIG. Interactions of age,
islet cell antibodies, insulin autoantibodies, and first-phase insulin response
in predicting risk of progression to IDDM in ICA+ relatives: the ICARUS data set. diab 1996;45:1720-1728.
59.
Ziegler AG, Ziegler R, Vardi P, Jackson
RA, Soeldner JS, Eisenbarth GS. Life-table analysis of progression to diabetes
of anti-insulin autoantibody-positive relatives of individuals with type I
diabetes. Diabetes 1989;38(10):1320-1325.
60.
Gianani R, Pugliese A, Bonner-Weir S et
al. Prognostically significant heterogeneity of cytoplasmic islet cell
antibodies in relatives of patients with type I diabetes. diab 1992;41(3):347-353.
61.
Marshall MO, Hoyer PE, Petersen JS et
al. Contribution of glutamate decarboxylase antibodies to the reactivity of
islet cell cytoplasmic antibodies. J Autoimmun 1994;7:497-508.
62.
Gianani R, Jackson R, Eisenbarth GS.
Evidence that the autoantigen of restricted ICA is GAD. Diabetes Res Clin Prac
14 (suppl. 1), S13. 1991.
Ref Type: Abstract
63.
Gianani R, Rabin DU, Verge CF et al.
ICA512 autoantibody radioassay. diab 1995;44(11):1340-1344.
64.
Steck AK, Zhang W, Bugawan TL et al. Do
non-HLA genes influence development of persistent islet autoimmunity and type 1
diabetes in children with high-risk HLA-DR,DQ genotypes? diab
2009;58(4):1028-1033.
65.
Noble JA, Valdes AM, Varney MD et al.
HLA class I and genetic susceptibility to type 1 diabetes: results from the
Type 1 Diabetes Genetics Consortium. diab 2010;59(11):2972-2979.
66.
Baschal EE, Aly TA, Babu SR et al.
HLA-DPB1*0402 Protects Against Type 1A Diabetic Autoimmunity in the Highest
Risk DR3-DQB1*0201/DR4-DQB1*0302 DAISY Population. diab 2007;56(9):2405-2409.
67.
Aly TA, Ide A, Jahromi MM et al. Extreme
Genetic Risk for Type 1A Diabetes. Proc Natl Acad Sci USA
2006;103(38):14074-14079.
68.
Winkler C, Krumsiek J, Lempainen J et
al. A strategy for combining minor genetic susceptibility genes to improve
prediction of disease in type 1 diabetes. Genes Immun 2012.
69.
Deschamps I, Lestradet H, Schmid M,
Busson M, Hors J. Risk to siblings of diabetic children: role of age and birth
order in relation to HLA-linked susceptibility. Pediatr Adolesc Endocrinol
1986;15:39-46.
70.
Lipton RB, LaPorte RE, Dorman JS, Riley
WJ, Trucco M, Becker DJ. A combination of HLA-DQ-beta non-Asp-57 homozygosity
and positive islet cell antybody (ICA) assay predicts insulin- dependent
diabetes in relatives of children with IDDM. Diabetes 40, 151a. 1991.
Ref Type: Abstract
71.
Pugliese A, Gianani R, Moromisato R et
al. HLA-DQB1*0602 is associated with dominant protection from diabetes even
among islet cell antibody-positive first-degree relatives of patients with
IDDM. diab 1995;44(6):608-613.
72.
Pugliese A, Kawasaki E, Zeller M et al.
Sequence analysis of the diabetes-protective human leukocyte antigen-DQB1*0602
allele in unaffected, islet cell antibody-positive first degree relatives and
in rare patients with type 1 diabetes. J Clin Endocrinol Metab
1999;84(5):1722-1728.
73.
Valdes AM, Thomson G, Graham J et al.
D6S265*15 marks a DRB1*15, DQB1*0602 haplotype associated with attenuated
protection from type 1 diabetes mellitus. diabetol 2005;48(12):2540-2543.
74.
Concannon P, Rich SS, Nepom GT. Genetics
of type 1A diabetes. N Engl J Med 2009;360(16):1646-1654.
75.
Hagopian WA, Erlich H, Lernmark A et al.
The Environmental Determinants of Diabetes in the Young (TEDDY): genetic
criteria and international diabetes risk screening of 421 000 infants. Pediatr
Diabetes 2011;12(8):733-743.
76.
Howson JM, Stevens H, Smyth DJ et al.
Evidence That HLA Class I and II Associations With Type 1 Diabetes,
Autoantibodies to GAD and Autoantibodies to IA-2, Are Distinct. diab
2011;60(10):2635-2644.
77.
Schatz D, Krischer J, Horne G et al.
Islet cell antibodies predict insulin-dependent diabetes in United States
school age children as powerfully as in unaffected relatives. J Clin Invest
1994;93(6):2403-2407.
78.
Bingley PJ, Bonifacio E, Shattock M et
al. Can islet cell antibodies predict IDDM in the general population? Diab care
1993;16:45-50.
79.
Kiviniemi M, Hermann R, Nurmi J et al. A
high-throughput population screening system for the estimation of genetic risk
for type 1 diabetes: an application for the TEDDY (the Environmental
Determinants of Diabetes in the Young) study. Diabetes Technol Ther
2007;9(5):460-472.
80.
Bonifacio E, Yu L, Williams AK et al.
Harmonization of glutamic acid decarboxylase and islet antigen-2 autoantibody
assays for national institute of diabetes and digestive and kidney diseases
consortia. J Clin Endocrinol Metab 2010;95(7):3360-3367.
81.
Yu L, Miao D, Scrimgeour L, Johnson K,
Rewers M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with
differential risk: nonradioactive bivalent proinsulin/insulin autoantibody
assay. diab 2012;61(1):179-186.
82.
Schlosser M, Mueller PW, Torn C,
Bonifacio E, Bingley PJ. Diabetes Antibody Standardization Program: evaluation
of assays for insulin autoantibodies. diabetol 2010;53(12):2611-2620.
83.
Kupila A, Muona P, Simell T et al.
Feasibility of genetic and immunological prediction of type I diabetes in a
population-based birth cohort. diabetol 2001;44(3):290-297.
84.
Rewers M, Norris JM, Eisenbarth GS et
al. Beta-cell autoantibodies in infants and toddlers without IDDM relatives:
Diabetes Autoimmunity Study in the Young (Daisy). J Autoimmun
1996;9(3):405-410.
85.
Schenker M, Hummel M, Ferber K et al.
Early expression and high prevalence of islet autoantibodies for DR3/4
heterozygous and DR4/4 homozygous offspring of parents with Type I diabetes:
the German BABYDIAB study. diabetol 1999;42(6):671-677.
86.
Virtanen SM, Laara E, Hypponen E et al.
Cow's milk consumption, HLA-DQB1 genotype, and type 1 diabetes: a nested
case-control study of siblings of children with diabetes. Childhood diabetes in
Finland study group [In Process Citation]. diab 2000;49(6):912-917.
87.
Achenbach P, Koczwara K, Knopff A,
Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to
(pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J
Clin Invest 2004;114(4):589-597.
88.
Achenbach P, Bonifacio E, Williams AJ,
Ziegler AG, Gale EA, Bingley PJ. Autoantibodies to IA-2beta improve diabetes
risk assessment in high-risk relatives. Diabetologia 2008;.
89.
Harjutsalo V, Podar T, Tuomilehto J.
Cumulative incidence of type 1 diabetes in 10,168 siblings of finnish
young-onset type 1 diabetic patients. diab 2005;54(2):563-569.
90.
Redondo MJ, Jeffrey J, Fain PR,
Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic
twins. N Engl J Med 2008;359(26):2849-2850.
91.
Gilliam LK, Brooks-Worrell BM, Palmer
JP, Greenbaum CJ, Pihoker C. Autoimmunity and clinical course in children with
type 1, type 2, and type 1.5 diabetes. J Autoimmun 2005;25(3):244-250.
92.
Bottazzo GF, Bosi E, Cull CA et al. IA-2
antibody prevalence and risk assessment of early insulin requirement in
subjects presenting with type 2 diabetes (UKPDS 71). diabetol
2005;48(4):703-708.
93.
Füchtenbusch M, Ferber K, Standl E,
Ziegler A-G, et al. Prediction of type I diabetes postpartum in patients with
gestational diabetes mellitus by combined islet cell autoantibody screening: A
prospective multicenter study. diab 1997;46:1459-1467.
94.
Diagnosis and Classification of Diabetes
Mellitus. Diab care 2005;28(suppl_1):S37-S42.
95.
Turner R, Stratton I, Horton V et al.
UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase
for prediction of insulin requirement in type 2 diabetes. UK Prospective
Diabetes Study Group. Lancet 1997;350(9087):1288-1293.
96.
Horton V, Stratton I, Bottazzo GF et al.
Genetic heterogeneity of autoimmune diabetes: age of presentation in adults is
influenced by HLA DRB1 and DQB1 genotypes (UKPDS 43). UK Prospective Diabetes
Study (UKPDS) Group. diabetol 1999;42(5):608-616.
97.
Maioli M, Pes GM, Delitala G et al.
Number of autoantibodies and HLA Genotype, more than high titers of glutamic
acid decarboxylase autoantibodies (GADA65Ab), predict insulin dependence in
latent autoimmune diabetes of adults (LADA). Eur J Endocrinol 2010.
98.
Rolandsson O, Palmer JP. Latent
autoimmune diabetes in adults (LADA) is dead: long live autoimmune diabetes! diabetol
2010;53(7):1250-1253.
99.
Naik RG, Brooks-Worrell BM, Palmer JP.
Latent autoimmune diabetes in adults. J Clin Endocrinol Metab
2009;94(12):4635-4644.
100.
Zimmet PZ, Tuomi T, Mackay IR et al.
Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to
glutamic acid decarboxylase in diagnosis and prediction of insulin dependency.
Diabet Med 1994;11(3):299-303.
101.
Seyfert-Margolis V, Gisler TD, Asare AL
et al. Analysis of T-cell assays to measure autoimmune responses in subjects
with type 1 diabetes: results of a blinded controlled study. Diabetes
2006;55(9):2588-2594.
102.
Fourlanos S, Dotta F, Greenbaum C et al.
Latent autoimmune diabetes in adults (LADA) should be less latent. diabetol
2005;48:2206-2212.
103.
Srikanta S, Ganda OP, Rabizadeh A,
Soeldner JS, Eisenbarth GS. First-degree relatives of patients with type I
diabetes mellitus. Islet cell antibodies and abnormal insulin secretion. N Engl
J Med 1985;313(8):461-464.
104.
Vardi P, Ziegler AG, Matthews JH et al.
Concentration of insulin autoantibodies at onset of type I diabetes. Inverse
log-linear correlation with age. Diab care 1988;11(9):736-739.
105.
Rewers M, LaPorte RE, King H, Tuomilehto
J. Trends in the prevalence and incidence of diabetes: insulin-dependent
diabetes mellitus in childhood. World Health Stat Q 1985;41(3-4):179-189.
106.
Laakso M, Pyorala K. Age at onset and
type of diabetes. Diab care 1985;8:114-117.
107.
Howson JM, Rosinger S, Smyth DJ, Boehm
BO, Todd JA.
Genetic analysis of adult-onset
autoimmune diabetes. diab 2011;60(10):2645-2653.
108.
Douek IF, Gillespie KM, Bingley PJ, Gale
EA. Diabetes in the parents of children with Type I diabetes. diabetol
2002;45(4):495-501.
109.
Japan IDDM Epidemiological Study Group.
Lack of regional variation in IDDM risk in Japan. Diab care 1993;16(5):796-800.
110.
Notsu K, Oka N, Note S, Nabeya N, Kuno
S, Sakurami T. Islet cell antibodies in the Japanese population and subjects
with type I (insulin-dependent) diabetes. diabetol 1985;28(9):660-662.
111.
Boehm BO, Manfras B, Seibler J et al.
Epidemiology and immunogenetic background of islet cell antibody positive
nondiabetic schoolchildren. diab 1991;40:1435-1439.
112.
Diabetes Epidemiology Research
International Group. Geographic patterns of childhood insulin-dependent
diabetes mellitus. diab 1988;37(8):1113-1119.
113.
Levy-Marchal C, Tichet J, Fajardy I, Gu
XF, Dubois F, Czernichow P. Islet cell antibodies in normal French
schoolchildren. diabetol 1992;35(6):577-582.
114.
Levy-Marchal C, Papoz L, de Beaufort C
et al. Incidence of juvenile type I (insulin-dependent) diabetes mellitus in
France. diabetol 1990;33(8):465-469.
115.
Bruining GJ, Molenaar JL, Grobbee DE et
al. Ten-year follow-up study of islet-cell antibodies and childhood diabetes
mellitus. Lancet 1989;1(8647):1100-1103.
116.
Green A, Gale EAM, Patterson CC.
Incidence of childhood-onset insulin-dependent diabetes mellitus: the EURODIAB
ACE Study. Lancet 1992;339(8798):905-909.
117.
Bergua M, Sole J, Marion G et al.
Prevalence of islet cell antibodies, insulin antibodies and hyperglycemia in
2291 schoolchildren. diabetol 1987;30(9):724-726.
118.
Serrano Rios M, Moy CS, Martin-Serrano R
et al. Incidence of type I (insulin-dependent) diabetes mellitus in subjects
0-14 years of age in the Comunidad of Madrid, Spain. diabetol
1990;33(7):422-424.
119.
Elliott RB, Pilcher CC, Stewart A,
Fergusson D, McGregor MA. The use of nicotinamide in the prevention of type I
diabetes. Ann N Y Acad Sci 1993;696:333-341.
120.
Patterson CC, Thorogood M, Smith PG,
Heasman MA, Clarke JA, Mann JI. Epidemiology of type I (insulin-dependent)
diabetes in scotland, 1968-1976: evidence of an increasing incidence. diabetol
1983;24:238-243.
121.
LaGasse JM, Brantley MS, Leech NJ et al.
Successful prospective prediction of type 1 diabetes in schoolchildren through
multiple defined autoantibodies: an 8-year follow-up of the Washington State
Diabetes Prediction Study. Diab care 2002;25(3):505-511.
122.
Dokheel TM, The Pittsburgh Epidemiology
Research Group. An epidemic of childhood diabetes in the United States? Diab
care 1993;16:1606-1611.
123.
Verge CF, Silink M, Howard NJ. The
incidence of childhood IDDM in New South Wales, Australia. Diab care
1994;17(7):693-696.
124.
Colman PG, McNair P, King J et al.
Screening for preclinical type 1 diabetes in a discrete population with an
apparent increased disease incidence. Pediatric Diabetes 2000;1:193-198.
125.
Bingley PJ, Gale EAM. Incidence of
insulin dependent diabetes in England: a study in the Oxford region, 1985-6.
BMJ 1989;298(6673):558-560.
126.
Dahlquist G, Blom L, Holmgren G et al.
The epidemiology of diabetes in Swedish children 0-14 years - a six year
prospective study. diabetol 1985;28(11):802-808.
127.
Karjalainen JK. Islet cell antibodies as
predictive markers for IDDM in children with high background incidence of
disease. diab 1990;39(9):1144-1150.
128.
Tuomilehto J, Lounamaa R,
Tuomilehto-Wolf E et al. Epidemiology of childhood diabetes mellitus in Finland
- background of a nationwide study of type I (insulin-dependent) diabetes
mellitus. diabetol 1992;35(1):70-76.
129.
Karjalainen J, Salmela P, Ilonen J,
Surcel H-M, Knip M. A comparison of childhood and adult type 1 diabetes
mellitus. New Engl J Med 1989;320:881-886.
130.
Tuomi T, Groop LC, Zimmet PZ, Rowley MJ,
Knowles W, Mackay IR. Antibodies to glutamic acid decarboxylase reveal latent
autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of
disease. diab 1993;42(2):359-362.
131.
Pilcher CC, Bibby NJ, Elliott RB.
Ontogeny of islet cell and insulin autoantibodies in first degree relatives of
IDDM. Autoimmunity 15, 78. 1993.
Ref Type: Abstract
132.
Ziegler AG, Hillebrand B, Rabl W et al.
On the appearance of islet associated autoimmunity in offspring of diabetic
mothers: a prospective study from birth. diabetol 1993;36:402-408.
133.
Naserke HE, Bonifacio E, Ziegler AG.
Prevalence, characteristics and diabetes risk associated with transient
maternally acquired islet antibodies and persistent islet antibodies in
offspring of parents with type 1 diabetes. J Clin Endocrinol Metab
2001;86(10):4826-4833.
134.
Vardi P, Soloveicik L, Barzilai D,
Giordanno-Galluzzo C, Seides Y, Perlman R. Insulin binding activity in cord
blood of normal pregnancies. Autoimmunity [15], 50. 1993.
Ref Type: Abstract
135.
Ziegler A-G, Hummel M, Schenker M,
Bonifacio E. Autoantibody appearance and risk for development of childhood
diabetes in offspring of parents with type 1 diabetes. The 2-year analysis of
the German BABYDIAB study. diab 1999;48:460-468.
136.
Baekkeskov S, Kanaani J, Jaume JC, Kash
S. Does GAD have a unique role in triggering IDDM? J Autoimmun
2000;15(3):279-286.
137.
Wegmann DR, Eisenbarth GS. It's Insulin.
J Autoimmun 2000;15(3):286-291.
138.
Vardi P, Dib SA, Tuttleman M et al.
Competitive insulin autoantibody assay. Prospective evaluation of subjects at
high risk for development of type I diabetes mellitus. diab
1987;36(11):1286-1291.
139.
Martino GV, Tappaz ML, Braghi S et al.
Autoantibodies to glutamic acid decarboxylase (GAD) detected by an
immuno-trapping enzyme activity assay: relation to insulin- dependent diabetes
mellitus and islet cell antibodies. J Autoimmun 1991;4(6):915-923.
140.
DeAizpurua HJ, Harrison LC, Cram DS. An
ELISA for antibodies to recombinant glutamic acid decarboxylase in IDDM. diab
1992;41(9):1182-1187.
141.
Thivolet CH, Tappaz M, Durand A et al.
Glutamic acid decarboxylase (GAD) autoantibodies are additional predictive
markers of Type 1 (insulin-dependent) diabetes mellitus in high risk
individuals. diabetol 1992;35:570-576.
142.
Sohnlein P, Muller M, Syren K et al.
Epitope spreading and a varying but not disease-specific GAD65 antibody
response in Type I diabetes. The Childhood Diabetes in Finland Study Group [In
Process Citation]. diabetol 2000;43(2):210-217.
143.
Hawa M, Fava D, Medici F et al.
Antibodies to IA-2 and GAD65 in type 1 and type 2 diabetes. Diab care
2000;23(2):228.
144.
Larsson L-I, Nielsen JH, Hutton JC,
Madsen OD. Pancreatic hormones are expressed on the surface of human and rat
islet cells through exocytotic sites. Eur J Cell Biol 1989;48(1):45-51.
145.
Aguilar-Diosdado M, Parkinson D, Corbett
JA et al. Potential autoantigens in IDDM. Expression of carboxypeptidase-H and
insulin but not glutamate decarboxylase on the beta-cell surface. diab
1994;43(3):418-425.
146.
Petersen JS, Russel S, Marshall MO et
al. Differential expression of glutamic acid decarboxylase in rat and human
islets. diab 1993;42:484-495.
147.
Sutherland DE, Sibley R, Xu XZ et al. Twin-to-twin
pancreas transplantation: reversal and reenactment of the pathogenesis of type
I diabetes. Trans Assoc Am Physicians 1984;97:80-87.
148.
Arslanian SA, Becker DJ, Rabin B et al.
Correlates of insulin antibodies in newly diagnosed children with
insulin-dependent diabetes before insulin therapy. diab 1985;34(9):926-930.
149.
Karjalainen J, Knip M, Mustonen A,
Ilonen J, Akerblom HK. Relation between insulin antibody and complement-fixing
islet cell antibody at clinical diagnosis of IDDM. diab 1986;35(5):620-622.
150.
Mcevoy RC, Witt ME, Ginsberg-Fellner F,
Rubinstein P. Anti-insulin antibodies in children with type I diabetes
mellitus. Genetic regulation of production and presence at diagnosis before
insulin replacement. diab 1986;35(6):634-641.
151.
Eisenbarth GS, Gianani R, Yu L et al.
Dual parameter model for prediction of type 1 diabetes mellitus. Proc Assoc Am
Physicians 1998;110(2):126-135.
152.
Castano L, Ziegler AG, Ziegler R,
Shoelson S, Eisenbarth GS. Characterization of insulin autoantibodies in
relatives of patients with type 1 diabetes. diab 1993;42:1202-1209.
153.
Williams AJK, Bingley PJ, Bonifacio E,
Palmer JP, Gale EAM. A novel micro-assay for insulin autoantibodies. J
Autoimmun 1997;10:473-478.
154.
Naserke HE, Dozio N, Ziegler A-G,
Bonifacio E. Comparison of a novel micro-assay for insulin autoantibodies with
the conventional radiobinding assay. diabetol 1998;41:681-683.
155.
Yu L, Robles DT, Abiru N et al. Early
expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence
for early determination of subsequent diabetes. Proc Natl Acad Sci USA
2000;97(4):1701-1706.
156.
Abiru N, Maniatis AK, Yu L et al.
Peptide and MHC specific breaking of humoral tolerance to native insulin with
the B:9-23 peptide in diabetes prone and normal mice. diab 2001;50:1274-1281.
157.
Moriyama H, Abiru N, Paronen J et al.
Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and
diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A 2003;100(18):10376-10381.
158.
Nakayama M, Abiru N, Moriyama H et al.
Prime role for an insulin epitope in the development of type 1 diabetes in NOD
mice. Nature 2005;435(7039):220-223.
159.
Crawford F, Stadinski B, Jin N et al.
Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes
in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A 2011.
160.
Stadinski BD, Zhang L, Crawford F,
Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin
bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S
A 2010;107(24):10978-10983.
161.
Pflueger M, Seppanen-Laakso T, Suortti T
et al. Age- and islet autoimmunity-associated differences in amino acid and
lipid metabolites in children at risk for type 1 diabetes. diab
2011;60(11):2740-2747.
162.
Oresic M, Simell S, Sysi-Aho M et al.
Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in
children who later progress to type 1 diabetes. J Exp Med
2008;205(13):2975-2984.
163.
Sysi-Aho M, Ermolov A, Gopalacharyulu PV
et al. Metabolic regulation in progression to autoimmune diabetes. PLoS Comput
Biol 2011;7(10):e1002257.
164.
Wang X, Jia S, Geoffrey R, Alemzadeh R,
Ghosh S, Hessner MJ. Identification of a Molecular Signature in Human Type 1
Diabetes Mellitus Using Serum and Functional Genomics. J Immunol
2008;180(3):1929-1937.
165.
Oak S, Gilliam LK, Landin-Olsson M et
al. The lack of anti-idiotypic antibodies, not the presence of the
corresponding autoantibodies to glutamate decarboxylase, defines type 1
diabetes. Proc Natl Acad Sci U S A 2008;.
166.
Ortqvist E, Brooks-Worrell B, Lynch K et
al. Changes in GAD65Ab-specific antiidiotypic antibody levels correlate with
changes in C-peptide levels and progression to islet cell autoimmunity. J Clin
Endocrinol Metab 2010;95(11):E310-E318.
167.
Hampe CS. Protective role of
anti-idiotypic antibodies in autoimmunity--lessons for type 1 diabetes.
Autoimmunity 2012;45(4):320-331.
168.
Soltesz G, Jeges S, Dahlquist G.
Non-genetic risk determinants for type 1 (insulin-dependent) diabetes mellitus
in childhood. Hungarian Childhood Diabetes Epidemiology Study Group. Acta
Paediatr 1994;83(7):730-735.
169.
Plagnol V, Howson JM, Smyth DJ et al.
Genome-wide association analysis of autoantibody positivity in type 1 diabetes
cases. PLoS Genet 2011;7(8):e1002216.
170.
Savilahti E, Simell O, Koskimies S,
Rilva A, Åkerblom HK. Celiac disease in insulin-dependent diabetes mellitus. J
Pediatr 1986;108(5 Pt 1):690-693.
171.
Maclaren NK, Riley WJ. Thyroid, gastric,
and adrenal autoimmunities associated with insulin-dependent diabetes mellitus.
Diab care 1985;8 (Suppl 1):34-38.
172.
Cotsapas C, Voight BF, Rossin E et al.
Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 2011;7(8):e1002254.
173.
Zhernakova A, Stahl EA, Trynka G et al.
Meta-analysis of genome-wide association studies in celiac disease and
rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet
2011;7(2):e1002004.
174.
Vang T, Congia M, Macis MD et al.
Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function
variant. Nat Genet 2005;37(12):1317-1319.
175.
Santin I, Castellanos-Rubio A, Aransay
AM, Castano L, Vitoria JC, Bilbao JR. The functional R620W variant of the
PTPN22 gene is associated with celiac disease. Tissue Antigens 2008;.
176.
van Oene M, Wintle RF, Liu X et al.
Association of the lymphoid tyrosine phosphatase R620W variant with rheumatoid
arthritis, but not Crohn's disease, in Canadian populations. Arthritis Rheum
2005;52(7):1993-1998.
177.
Soltesz G, Jeges S, Dahlquist G,
Hungarian Childhood Diabetes Epidemiology Study Group. Non-genetic risk
determinants for Type 1 (insulin-dependent) diabetes. Acta Paediatr
1994;83:730-735.
178.
Gillespie KM, Bain SC, Barnett AH et al.
The rising incidence of childhood type 1 diabetes and reduced contribution of
high-risk HLA haplotypes. Lancet 2004;364(9446):1699-1700.
179.
Kronenberg M, Siu G, Hood LE, Shastri N.
The molecular genetics of the T-cell antigen receptor and T-cell antigen
recognition. Ann Rev Immunol 1986;4:529-591.
180.
Joner G, Sovik O. Increasing incidence
of diabetes mellitus in norwegian children 0-14 years of age, 1973-1982.
diabetol 1989;32:79-83.
181.
Green A, Andersen PK, Svendsen AJ,
Mortensen K. Increasing incidence of early onset type I (insulin-dependent)
diabetes mellitus: a study of Danish male birth cohorts. diabetol
1992;35:178-182.
182.
Soltesz G, Madacsy L, Bekefi D, Danko I.
Rising incidence of type 1 diabetes in Hungarian children (1978- 1987).
Hungarian Childhood Diabetes Epidemiology Group. Diabetic Med 1990;7:111-114.
183.
Tuomilehto J, Rewers M, Reunanen A et
al. Increasing trend in type I (insulin-dependent) diabetes mellitus in
childhood in Finland. Analysis of age, calendar time, and birth cohort effects
during 1965 to 1984. diabetol 1991;34(4):282-287.
184.
Drykoningen CEM, Mulder ALM, Vaandrager
GJ, LaPorte RE, Bruining GJ. The incidence of male childhood type 1
(insulin-dependent) diabetes mellitus is rising rapidly in the Netherlands.
diabetol 1992;35(2):139-142.
185.
Nystrom L, Dahlquist G, Rewers M, Wall
S. The Swedish childhood diabetes study: an analysis of the temporal variation
in diabetes incidence, 1978-1987. Int J Epidemiol 1990;19:141-146.
186.
Rewers M, LaPorte RE, Walczak M,
Dmochowski K, Bogaczynska E. Apparent epidemic of insulin dependent diabetes
mellitus in Midwestern Poland. diab 1987;36(1):106-113.
187.
Hayashi T, Faustman D. Essential role of
human leukocyte antigen-encoded proteasome subunits in NF-kappaB activation and
prevention of tumor necrosis factor-alpha-induced apoptosis. J Biol Chem
2000;275(7):5238-5247.
188.
Onkamo P, Vaananen S, Karvonen M,
Tuomilehto J. Worldwide increase in incidence of Type I diabetes--the analysis
of the data on published incidence trends. diabetol 1999;42(12):1395-1403.
189.
Eurodiab ACE Study Group. Variation and
trends in incidence of childhood diabetes in Europe. EURODIAB ACE Study Group.
Lancet 2000;355(9207):873-876.
190.
Elliott RB, Pilcher C, Edgar BW.
Geographic IDDM in Polynesia and Macronesia: the epidemiology of insulin
dependent diabetes in Polynesian children born and reared in Polynesia,
compared with Polynesian children resident in Auckland, New Zealand. Diabetes
in the Young Bulletin (Proc of the ISGD) 20, 16. 1989.
Ref Type: Abstract
191.
Bodansky HJ, Staines A, Stephenson C,
Haigh D, Cartwright R. Evidence for an environmental effect in the aetiology of
insulin dependent diabetes in a transmigratory population. BMJ
1992;304(6833):1020-1022.
192.
Kostraba JN, Gay EC, Cai Y et al.
Incidence of insulin-dependent diabetes mellitus in Colorado. Epidemiology
1992;3:232-238.
193.
Menser MA, Forrest JM, Bransby RD.
Rubella infection and diabetes mellitus. Lancet 1978;1(8055):57-60.
194.
Ginsberg-Fellner F, Dobersen MJ, Witt
ME, Rayfield EJ, Rubinstein P, Notkins AL. HLA antigens, cytoplasmic islet cell
antibodies, and carbohydrate tolerance in families of children with
insulin-dependent diabetes mellitus. diab 1982;31:292-298.
195.
Clarke WL, Shaver KA, Bright GM, Rogol
AD, Nance WE. Autoimmunity in congenital rubella syndrome. J Pediatr
1984;104(3):370-373.
196.
Viskari H, Paronen J, Keskinen P et al.
Humoral beta-cell autoimmunity is rare in patients with the congenital rubella
syndrome. Clin Exp Immunol 2003;133(3):378-383.
197.
Stene LC, Rewers M. Immunology in the
clinic review series; focus on type 1 diabetes and viruses: the enterovirus
link to type 1 diabetes: critical review of human studies. Clin Exp Immunol
2012;168(1):12-23.
198.
Stene LC, Oikarinen S, Hyoty H et al.
Enterovirus infection and progression from islet autoimmunity to type 1
diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). diab
2010;59(12):3174-3180.
199.
Yoon J-W, Morishima T, McClintock PR, Austin
M, Notkins AL. Virus-induced diabetes mellitus: mengovirus infects pancreatic
beta cells in strains of mice resistant to the diabetogenic effect of
encephalomyocarditis virus. J Virol 50, 684-690. 1984.
Ref Type: Generic
200.
Tuvemo T, Dahlquist G, Frisk G et al.
The Swedish childhood diabetes study III:
IgM against coxsackie B viruses in newly diagnosed type 1
(insulin-dependent) diabetic children - no evidence of increased antibody
frequency. diabetol 1989;32(10):745-747.
201.
Kaufman DL, Erlander MG, Clare-Salzler
M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate
decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest
1992;89(1):283-292.
202.
Karounos DG, Simmerman L, Hickman SL,
Jacob RJ. Identification of the p52-Rubella related autoantigen as an insulin
secretory granule protein. Diabetes 42 (Suppl 1), 221A. 1993.
Ref Type: Abstract
203.
Lack of regional variation in IDDM risk
in Japan. Japan IDDM Epidemiology Study Group. Diab care 1993;16(5):796-800.
204.
Kawasaki E, Eguchi K. Is Type 1 diabetes
in the Japanese population the same as among Caucasians? Ann N Y Acad Sci
2004;1037:96-103.:96-103.
205.
Hanafusa T, Imagawa A. Insulitis in
human type 1 diabetes. Ann N Y Acad Sci 2008;1150:297-299.
206.
Brown RJ, Rother KI, Artman H et al.
Minocycline-induced drug hypersensitivity syndrome followed by multiple
autoimmune sequelae. Arch Dermatol 2009;145(1):63-66.
207.
Sekine N, Motokura T, Oki T et al. Rapid
loss of insulin secretion in a patient with fulminant type 1 diabetes mellitus
and carbamazepine hypersensitivity syndrome. JAMA 2001;285(9):1153-1154.
208.
Ostrov DA, Grant BJ, Pompeu YA et al.
Drug hypersensitivity caused by alteration of the MHC-presented self-peptide
repertoire. Proc Natl Acad Sci U S A 2012;109(25):9959-9964.
209.
Illing PT, Vivian JP, Dudek NL et al.
Immune self-reactivity triggered by drug-modified HLA-peptide repertoire.
Nature 2012;486(7404):554-558.
210.
Dahlquist GG. Primary and secondary
prevention strategies of pre-type 1 diabetes. Potentials and pitfalls. [Review]
[27 refs]. Diab care 1999;22 Suppl 2:B4-B6.
211.
Lonnrot M, Korpela K, Knip M et al.
Enterovirus infection as a risk factor for beta-cell autoimmunity in a
prospectively observed birth cohort: the Finnish Diabetes Prediction and
Prevention Study. diab 2000;49(8):1314-1318.
212.
Akerblom HK, Virtanen SM, Ilonen J et
al. Dietary manipulation of beta cell autoimmunity in infants at increased risk
of type 1 diabetes: a pilot study. diabetol 2005;48(5):829-837.
213.
Norris JM, Beaty B, Klingensmith G et
al. Lack of association between early exposure to cow's milk protein and b-cell autoimmunity: Diabetes
Autoimmunity Study in the Young (DAISY). JAMA 1996;276:609-614.
214.
Graves PM, Rewers M. The role of
enteroviral infections in the development of IDDM: limitations of current
approaches. diab 1997;46(2):161-168.
215.
Graves PM, Barriga KJ, Norris JM et al.
Lack of association between early childhood immunizations and beta-cell
autoimmunity. Diab care 1999;22(10):1694-1697.
216.
Honeyman MC, Coulson BS, Stone NL et al.
Association between rotavirus infection and pancreatic islet autoimmunity in
children at risk of developing type 1 diabetes. diab 2000;49(8):1319-1324.
217.
Blomqvist M, Juhela S, Erkkila S et al.
Rotavirus infections and development of diabetes-associated autoantibodies
during the first 2 years of life. Clin Exp Immunol 2002;128(3):511-515.
218.
Medzhitov R, Preston-Hurlburt P, Janeway
CA. A human homologue of the Drosophila Toll protein signals activation of
adaptive immunity. Nature 1997;388(6640):394-397.
219.
Devendra D, Jasinski J, Melanitou E et
al. Interferon-{alpha} as a Mediator of Polyinosinic:Polycytidylic Acid-Induced
Type 1 Diabetes. diab 2005;54(9):2549-2556.
220.
Zipris D, Lien E, Nair A et al.
TLR9-signaling pathways are involved in Kilham rat virus-induced autoimmune
diabetes in the biobreeding diabetes-resistant rat. J Immunol
2007;178(2):693-701.
221.
Londono P, Komura A, Hara N, Zipris D.
Brief dexamethasone treatment during acute infection prevents virus-induced
autoimmune diabetes. Clin Immunol 2010;135(3):401-411.
222.
Sobel DO, Goyal D, Ahvazi B et al. Low
dose poly I:C prevents diabetes in the diabetes prone BB rat. Journal of
Autoimmunity 1998;11:343-352.
223.
Wen L, Ley RE, Volchkov PY et al. Innate
immunity and intestinal microbiota in the development of Type 1 diabetes.
Nature 2008;455(7216):1109-1113.
224.
Wilberz S, Partke HJ, Dagnaes-Hansen F,
Herberg L. Persistent MHV (mouse hepatitis virus) infection reduces the
incidence of diabetes mellitus in non-obese diabetic mice. diabetol
1991;34(1):2-5.
225.
Oldstone MB. Prevention of type I
diabetes in nonobese diabetic mice by virus infection. Science
1988;239:500-502.
226.
Dyrberg T, Schwimmbeck PL, Oldstone MB.
Inhibition of diabetes in BB rats by virus infection. J Clin Invest
1988;81(3):928-931.
227.
Guberski DL, Thomas VA, Shek WR et al.
Induction of Type 1 diabetes by Kilham's rat virus in diabetes-resistant BB/Wor
rats. Science 1991;254:1010-1013.
228.
Zipris D, Lien E, Xie JX, Greiner DL,
Mordes JP, Rossini AA. TLR activation synergizes with Kilham rat virus
infection to induce diabetes in BBDR rats. J Immunol 2005;174(1):131-142.
229.
Yoon JW. Role of viruses in the
pathogenesis of IDDM. Ann Med 23, 437-445. 1991.
Ref Type: Generic
230.
Horwitz MS, Fine C, Ilic A, Sarvetnick
N. Requirements for Viral-mediated Autoimmune Diabetes: beta-Cell Damage and
Immune Infiltration. J Autoimmun 2001;16(3):211-217.
231.
Yoon J-W, McClintock PR, Bachurski CJ,
Longstreth JD, Notkins AL. Virus-induced diabetes mellitus. No evidence for
immune mechanisms in the destruction of beta-cells by the D variant of
encephalyomyocarditis virus. diab 1985;34(9):922-925.
232.
Virtanen S, Rasanen L, Aro A et al.
Infant feeding in Finnish Children less than 7 yr of age with newly diagnosed
IDDM. Childhood Diabetes in Finland Study Group. Diab care 1991;14(5):415-417.
233.
Virtanen SM, Rasanen L, Aro A et al.
Feeding in infancy and the risk of type 1 diabetes mellitus in Finnish
children. The 'Childhood Diabetes in Finland' Study Group. Diabetic Med
1992;9(9):815-819.
234.
Mayer EJ, Hamman RF, Gay EC, Lezotle DC,
Savitz DA, Klingensmith GJ. Reduced risk of IDDM among breast-fed children. The
Colorado IDDM Registry. diab 1988;37(12):1625-1632.
235.
Verge CF, Howard NJ, Irwig L, Simpson
JM, Mackerras D, Silink M. Environmental factors in childhood IDDM. A
population-based, case-control study. Diab care 1994;17(12):1381-1389.
236.
Kyvik KO, Green A, Svendsen A, Mortensen
K. Breast feeding and the development of type I diabetes mellitus. Diabetic Med
1992;9(3):233-235.
237.
Gerstein HC. Cow's milk exposure and
type I diabetes mellitus - a critical overview of the clinical literature. Diab
care 1994;17:13-19.
238.
Johansson C, Samuelsson U, Ludvigsson J.
A high weight gain early in life is associated with an increased risk of type I
(insulin-dependent) diabetes mellitus. diabetol 1994;37(1):91-94.
239.
Savilahti E, Akerblom HK, Tainio V-M,
Koshkimies S. Children with newly diagnosed insulin dependent diabetes mellitus
have increased levels of cow's milk antibodies. Diabetes Res 1988;7(3):137-140.
240.
Yokota A, Yamaguchi Y, Ueda Y et al.
Comparison of islet cell antibodies, islet cell surface antibodies and
anti-bovine serum albumin antibodies in type 1 diabetes. Diabetes Res Clin Prac
1990;9(3):211-217.
241.
Pocecco M, Nicoloso F, Tonini G, Presani
G, Marinoni S. Increased levels of cow's milk antibodies in children with newly
diagnosed insulin-dependent diabetes mellitus (IDDM). Horm Metab Res 35, 67.
1991.
Ref Type: Abstract
242.
Dahlquist G, Savilahti E, Landin-Olson
M. An increased level of antibodies to beta-lactoglobulin is a risk determinant
of early-onset type I (insulin-dependent) diabetes mellitus independent of
islet cell antibodies and early introduction of cow's milk. diabetol
1992;35(10):980-984.
243.
Karjalainen J, Saukkonen T, Savilahti E,
Dosch H-M. Disease-associated anti-bovine serum albumin antibodies in type I
(insulin-dependent) diabetes mellitus are detected by particle concentration
fluoroimmunoassay, and not by enzyme linked immunoassay. diabetol
1992;35(10):985-990.
244.
Atkinson MA, Bowman MA, Kao K et al.
Lack of immune responsiveness to bovine serum albumin in insulin- dependent
diabetes. N Engl J Med 1993;329:1853-1858.
245.
Elliott RB, Martin JM. Dietary protein:
a trigger of insulin-dependent diabetes in the BB rat? diabetol
1984;26(4):297-299.
246.
Daneman D, Fishman L, Clarson C, Martin
JM. Dietary triggers of insulin-dependent diabetes in the BB rat. Diabetes Res
1987;5(2):93-97.
247.
Coleman DL, Kuzava JE, Leiter EH. Effect
of diet on incidence of diabetes in nonobese diabetic mice. diab
1990;39:432-436.
248.
Elliott RB, Reddy SN, Bibby NJ, Kida K.
Dietary prevention of diabetes in the non-obese diabetic mouse. diabetol
1988;31(1):62-64.
249.
Paronen J, Knip M, Savilahti E et al.
Effect of cow's milk exposure and maternal type 1 diabetes on cellular and
humoral immunization to dietary insulin in infants at genetic risk for type 1
diabetes. Finnish Trial to Reduce IDDM in the Genetically at Risk Study Group.
diab 2000;49(10):1657-1665.
250.
Vaarala O, Ilonen J, Ruohtula T et al.
Removal of Bovine Insulin From Cow's Milk Formula and Early Initiation of
Beta-Cell Autoimmunity in the FINDIA Pilot Study. Arch Pediatr Adolesc Med
2012.
251.
Norris JM, Barriga K, Klingensmith G et
al. Timing of cereal exposure in infancy and risk of islet autoimmunity. The Diabetes Autoimmunity Study in the Young
(DAISY). JAMA 2003;290(13):1713-1720.
252.
Ziegler AG, Schmid S, Huber D, Hummel M,
Bonifacio E. Early infant feeding and risk of developing type 1
diabetes-associated autoantibodies. JAMA 2003;290(13):1721-1728.
253.
Norris JM, Yin X, Lamb MM et al. Omega-3
polyunsaturated fatty acid intake and islet autoimmunity in children at
increased risk for type 1 diabetes. JAMA 2007;298(12):1420-1428.
254.
Miller MR, Yin X, Seifert J et al.
Erythrocyte membrane omega-3 fatty acid levels and omega-3 fatty acid intake are
not associated with conversion to type 1 diabetes in children with islet
autoimmunity: The Diabetes Autoimmunity Study in the Young (DAISY). Pediatr
Diabetes 2011.
255.
Deane KD, Striebich CC, Goldstein BL et
al. Identification of undiagnosed inflammatory arthritis in a community health
fair screen. Arthritis Rheum 2009;61(12):1642-1649.
256.
Srikanta S, Ganda OP, Eisenbarth GS,
Soeldner JS. Islet cell antibodies and beta cell function in monozygotic
triplets and twins initially discordant for Type I diabetes mellitus. N Engl J
Med 1983;308(6):322-325.
257.
Soeldner JS, Tuttleman M, Srikanta S,
Ganda OP, Eisenbarth GS. Insulin-dependent diabetes mellitus autoimmunity:
islet-cell autoantibodies,insulin autoantibodies and beta-cell failure. N Engl
J Med 1985;313(14):893-894.
258.
Eisenbarth GS. Type I diabetes
mellitus. A chronic autoimmune disease.
N Engl J Med 1986;314:1360-1368.
259.
Bleich D, Jackson RA, Soeldner JS,
Eisenbarth GS. Analysis of metabolic progression to type I diabetes in ICA+ relatives
of patients with type I diabetes. Diab care 1990;13(2):111-118.
260.
Ziegler AG, Schmid S, Huber D, Hummel M,
Bonifacio E. Early infant feeding and risk of developing type 1
diabetes-associated autoantibodies. Journal of the American Medical Association
2003;290(13):1721-1728.
261.
McCulloch DK, Klaff LJ, Kahn SE et al.
Nonprogression of subclinical B-cell dysfunction among first degree relatives
of IDDM patients. 5 year follow-up of
the Seattle Family Study. diab 1990;39(5):549-556.
262.
Beer SF, Heaton DA, Alberti KG, Pyke DA,
Leslie RD. Impaired glucose tolerance precedes but does not predict
insulin-depedendent diabetes mellitus: a study of identical twins. diabetol
1990;33(8):497-502.
263.
Kobayashi T, Itoh T, Kosaka K, Sato K,
Tsuji K. Time course of islet cell antibodies and beta-cell function in
non-insulin-dependent stage of type I diabetes. diab 1987;36(4):510-517.
264.
Tunbridge WMG, Brewis M, French JM et
al. Natural history of autoimmune thyroiditis. BMJ 1981;282(6260):258-262.
265.
Spencer KM, Tarn A, Dean BM, Lister J,
Bottazzo GF. Fluctuating islet-cell autoimmunity in unaffected relatives of
patients with insulin-dependent diabetes. Lancet 1984;1(8380):764-766.
266.
Chase HP, Voss MA, Butler-Simon N, Hoops
S, O'Brien D, Dobersen MJ. Diagnosis of pre-type I diabetes. J Pediatr
1987;111(6 Pt 1):807-812.
267.
Thivolet C, Beaufrere B, Geburher L,
Chatelain P, Orgiazzi J, Francois R. Autoantibodies and genetic factors
associated with the development of type I (insulin-dependent) diabetes in first
degree relatives of diabetic patients. diabetol 1991;34(3):186-191.
268.
Helmke K, Otten A, Willems WR et al.
Islet cell antibodies and the development of diabetes mellitus in relation to
mumps infection and mumps vaccination. diabetol 1986;29(1):30-33.
269.
Landin-Olsson M, Karlsson A, Dahlquist G
et al. Islet cell and other organ-specific autoantibodies in all children
developing Type 1 (insulin-dependent) diabetes mellitus in Sweden during one
year and in matched control children. diabetol 1989;32:387-395.
270.
Landin-Olsson M. Precision of the
islet-cell antibody assay depends on the pancreas. J Clin Lab Anal
1990;4:289-294.
271.
Kulmala P, Rahko J, Savola K et al.
Stability of autoantibodies and their relation to genetic and metabolic markers
of Type I diabetes in initially unaffected schoolchildren. diabetol
2000;43(4):457-464.
272.
Yu J, Yu L, Bugawan TL et al. Transient
anti-islet autoantibodies: infrequent occurrence and lack of association with
genetic risk factors. J Clin Endocrinol Metab 2000;85(7):2421-2428.
273.
Colman PG, Steele C, Couper JJ et al.
Islet autoimmunity in infants with a Type I diabetic relative is common but is
frequently restricted to one autoantibody. diabetol 2000;43(2):203-209.
274.
Barker JM, Barriga K, Yu L et al.
Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young
(DAISY). J Clin Endocrinol Metab 2004;89:3896-3902.
275.
Douek IF, Leech NJ, Bingley PJ, Gale EA.
Eczema and Type 1 diabetes. Diabet Med 2002;19(2):174-175.
276.
Achenbach P, Warncke K, Reiter J et al.
Stratification of type 1 diabetes risk on the basis of islet autoantibody
characteristics. diab 2004;53(2):384-392.
277.
Carel JC, Boitard C, Bougneres PF. Decreased
insulin response to glucose in islet cell antibody- negative siblings of type 1
diabetic children. J Clin Invest 1993;92(1):509-513.
278.
Pilcher CC, Elliott RB. A sensitive and
reproducible method for the assay of human islet cell antibodies. J Immunol
Methods 1990;129(1):111-117.
279.
McCulloch DK, Koerker DJ, Kahn SE,
Bonner-Weir S, Palmer JP. Correlations of in vivo beta-cell function tests with
beta-cell mass and pancreatic insulin content in streptozocin-treated baboons.
diab 1991;40(6):673-679.
280.
Allen HF, Jeffers BW, Klingensmith GJ,
Chase HP. First-phase insulin release in normal children. J Pediatr
1993;123(5):733-738.
281.
Arslanian S, Austin A. Determinants of
first and second phase insulin secretion in healthy adolescents. Pediatr Res
3[Suppl], S74. 1993.
Ref Type: Abstract
282.
Cutfield WS, Bergman RN, Menon RK,
Sperling MA. The modified minimal model: application to measurement of insulin
sensitivity in children. J Clin Endocrinol Metab 1990;70(6):1644-1650.
283.
Smith CP, Williams AJK, Thomas JM et al.
The pattern of basal and stimulated insulin responses to intravenous glucose in
first degree relatives of type I (insulin-dependent) diabetic children and
unrelated adults aged 5 to 50 years. diabetol 1988;31(7):430-434.
284.
Colman PG, Stewart V, Kean J et al.
Comparison of two commonly used standard IVGTTs. Diab care
1992;15(8):1053-1055.
285.
Bingley PJ, Colman P, Eisenbarth GS et
al. Standardization of IVGTT to predict IDDM. Diab care 1992;15:1313-1316.
286.
Rayman G, Clark P, Schneider AE, Hales
CN. The first phase insulin response to intravenous glucose is highly
reproducible. diabetol 1990;33:631-634.
287.
Smith CP, Tarn AC, Thomas JM et al.
Between and within subject variation of the first phase insulin response to
intravenous glucose. diabetol 1988;31(2):123-125.
288.
McNair PD, Colman PG, Alford FP,
Harrison LC. Reproducibility of the first phase insulin response (FPIR) in the
intravenous glucose tolerance test (IVGTT) is not improved by retrograde
cannulation and arterialisation or by the use of a lower glucose dose.
Autoimmunity . 1993.
Ref Type: Abstract
289.
Rowe RE, Leech NJ, McCullock DK. Does
drawing blood from a retrogradely cannulated hand vein improve reproducibility
in the intravenous glucose tolerance test (IVGTT)? Autoimmunity 15 (Suppl), 79.
1993.
Ref Type: Abstract
290.
Stene LC, Barriga K, Hoffman M et al.
Normal but increasing hemoglobin A1c levels predict progression from islet
autoimmunity to overt type 1 diabetes: Diabetes Autoimmunity Study in the Young
(DAISY). Pediatr Diabetes 2006;7(5):247-253.
291.
Vehik K, Cuthbertson D, Boulware D et
al. Performance of HbA1c as an Early Diagnostic Indicator of Type 1 Diabetes in
Children and Youth. Diab care 2012;35(9):1821-1825.
292.
Leech NJ, Rowe RE, Bucksa J, McCulloch
DK. HbA1c may be an early indicator of islet cell dysfunction. Autoimmunity 15
(Suppl), 75. 1993.
Ref Type: Abstract
293.
Santiago JV. Insulin therapy in the last
decade. A pediatric perspective. Diab care 1993;16 (Suppl 3):143-154.
294.
Signore A, Chianelli M, Ferretti E,
Multari G, Andreani D, Pozzilli P. New tools for imaging insulitis in pre
diabetes. Autoimmunity 15(Suppl.), 64. 1993.
Ref Type: Abstract
295.
Yuh WTC, Wiese JA, Abu-Yousef MM et al.
Pancreatic transplant imaging. Radiology 1988;167(3):679-683.
296.
Kelcz F, Sollinger HW, Pirsch JD. MRI of
the pancreas transplant: lack of correlation between imaging and clinical
status. Mag Res Med 1991;21(1):30-38.
297.
Fernandez MP, Bernardino ME, Neylan JF,
Olson RA. Diagnosis of pancreatic transplant dysfunction: value of
gadopentetate dimeglumine-enhanced MR imaging. AJR 1991;156(6):1171-1176.
298.
Turvey SE, Swart E, Denis MC et al.
Noninvasive imaging of pancreatic inflammation and its reversal in type 1
diabetes. J Clin Invest 2005;115(9):2454-2461.
299.
Moore A, Grimm J, Han B, Santamaria P.
Tracking the recruitment of diabetogenic CD8+ T-cells to the pancreas in real
time. diab 2004;53(6):1459-1466.
300.
Atkinson MA, Gianani R. The pancreas in
human type 1 diabetes: providing new answers to age-old questions. Curr Opin
Endocrinol Diabetes Obes 2009;16(4):279-285.
301.
Souza F, Simpson N, Raffo A et al.
Longitudinal noninvasive PET-based beta cell mass estimates in a spontaneous
diabetes rat model. J Clin Invest 2006;116(6):1506-1513.
302.
Hanafusa T, Miyazaki A, Miyagawa J et
al. Examination of islets in the pancreas biopsy specimens from newly diagnosed
type I (insulin-dependent) diabetic patients. diabetol 1990;33:105-111.
303.
Shimada A, Morimoto J, Kodama K et al.
T-cell-mediated autoimmunity may be involved in fulminant type 1 diabetes. Diab
care 2002;25(3):635-636.
304.
Abiru N, Kawasaki E, Eguch K. Current
knowledge of Japanese type 1 diabetic syndrome. Diabetes Metab Res Rev
2002;18(5):357-366.
305.
Tiberti C, Buzzetti R, Anastasi E et al.
Autoantibody negative new onset type 1 diabetic patients lacking high risk HLA
alleles in a caucasian population: are these type 1b diabetes cases? [In
Process Citation]. Diabetes Metab Res Rev 2000;16(1):8-14.
306.
Gianani R, Putnam A, Still T et al.
Initial results of screening of non - diabetic organ donors for expression of
islet autoantibodies. J Clin Endocrinol Metab 2006;91:1855-1861.
307.
Campbell-Thompson M, Wasserfall C,
Kaddis J et al. Network for Pancreatic Organ Donors with Diabetes (nPOD):
developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 2012.
308.
Gianani R, Campbell-Thompson M, Sarkar
SA et al. Dimorphic histopathology of long-standing childhood-onset diabetes.
diabetol 2010.
309.
Gale EA. To boldly go-or to go too
boldly? The accelerator hypothesis revisited. Diabetologia
2007;50(8):1571-1575.
310.
Wilkin TJ. The accelerator hypothesis:
weight gain as the missing link between Type I and Type II diabetes. diabetol
2001;44(7):914-922.
311.
Kibirige M, Metcalf B, Renuka R, Wilkin
TJ. Testing the accelerator hypothesis: the relationship between body mass and
age at diagnosis of type 1 diabetes. Diabetes Care 2003;26(10):2865-2870.
312.
Pundziute-Lycka A, Persson LA, Cedermark
G et al. Diet, growth, and the risk for type 1 diabetes in childhood: a matched
case-referent study. Diabetes Care 2004;27(12):2784-2789.
313.
Bingley PJ, Mahon JL, Gale EA. Insulin
resistance and progression to type 1 diabetes in the European Nicotinamide
Diabetes Intervention Trial (ENDIT). Diabetes Care 2008;31(1):146-150.
314.
Martin S, Pawlowski B, Greulich B,
Ziegler AG, Mandrup-Poulsen T, Mahon J. Natural course of remission in IDDM
during 1st year after diagnosis. Diab care 1992;15(1):66-74.
315.
Schiffrin A, Suissa S, Weitzner G,
Poussier P, Lalla D. Factors predicting course of B-cell function in IDDM. Diab
care 1992;15(8):997-1001.
316.
Knip M, Ilonen J, Mustonen A, Akerblom
HK. Evidence for an accelerated B-cell destruction in HLA-Dw3/Dw4 heterozygous
children with type I (insulin-dependent) diabetes. diabetol 1986;29(6):347-351.
317.
Karges B, Durinovic-Bello I, Heinze E,
Boehm BO, Debatin KM, Karges W. Complete long-term recovery of beta-cell
function in autoimmune type 1 diabetes after insulin treatment. Diabetes Care
2004;27(5):1207-1208.
318.
Baisch JM, Weeks T, Giles R, Hoover M,
Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in
insulin- dependent diabetes mellitus. N Engl J Med 1990;322(26):1836-1841.
319.
Erlich HA, Griffith RL, Bugawan TL,
Ziegler R, Alper C, Eisenbarth GS. Implication of specific DQB1 alleles in
genetic susceptibility and resistance by identification of IDDM siblings with
novel HLA-DQB1 allele and unusual DR2 and DR1 haplotypes. diab
1991;40(4):478-481.
320.
Shields BM, McDonald TJ, Ellard S,
Campbell MJ, Hyde C, Hattersley AT. The development and validation of a
clinical prediction model to determine the probability of MODY in patients with
young-onset diabetes. diabetol 2012.
321.
Hattersley AT, Ashcroft FM. Activating
mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new
scientific insights, and new therapy. diab 2005;54(9):2503-2513.
322.
Karasik A, O'Hara C, Srikanta S, Swift
M, et al. Genetically programmed selective islet beta-cell loss in diabetic
subjects with Wolfram's syndrome. Diab care 1989;12:135-138.
323.
Kadowaki T, Kadowaki H, Mori Y et al. A
subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N
Engl J Med 1994;330(14):962-968.
324.
Bowden DW, Gravius TC, Akots G, Fajans
SS. Identification of genetic markers flanking the locus for maturity-onset
diabetes of the young on human chromosome 20. diab 1992;41:88-92.
325.
Palmer JP, Hampe CS, Chiu H, Goel A,
Brooks-Worrell BM. Is latent autoimmune diabetes in adults distinct from type 1
diabetes or just type 1 diabetes at an older age? Diabetes 2005;54 Suppl
2:S62-7.:S62-S67.
326.
Carlsson A, Sundkvist G, Groop L, Tuomi
T. Insulin and glucagon secretion in patients with slowly progressing
autoimmune diabetes (LADA). J Clin Endocrinol Metab 2000 Jan ;85 (1 ):76 -80
2000;85(1):76-80.
327.
Herskowitz-Dumont R, Wolfsdorf JI,
Jackson RA, Eisenbarth GS. Distinction between transient hyperglycemia and
early insulin-dependent diabetes mellitus in childhood: a prospective study of
incidence and prognostic factors. J Pediatr 1993;123:347-354.
328.
Gale EA, Bingley PJ, Emmett CL, Collier
T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled
trial of intervention before the onset of type 1 diabetes. Lancet
2004;363(9413):925-931.
329.
Effects of insulin in relatives of
patients with type 1 diabetes mellitus. N Engl J Med 2002;346(22):1685-1691.
330.
Trudeau JD, Kelly-Smith C, Verchere CB
et al. Prediction of spontaneous autoimmune diabetes in NOD mice by
quantification of autoreactive T cells in peripheral blood. J Clin Invest
2003;111(2):217-223.
331.
Lieberman SM, Evans AM, Han B et al.
Identification of the {beta} cell antigen targeted by a prevalent population of
pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A
2003;100(14):8384-8388.
332.
Hutton JC, Eisenbarth GS. A pancreatic
{beta}-cell-specific homolog of glucose-6-phosphatase emerges as a major target
of cell-mediated autoimmunity in diabetes. Proc Natl Acad Sci U S A
2003;100:8626-8628.