At present we can predict the development of type 1A diabetes but do not have a safe and effective preventive therapy (1;2). One can estimate from the presence of multiple anti-islet autoantibodies in approximately 1/300 individuals in the general population that approximately one million individuals in the United States are currently at high risk of developing type 1 diabetes and thus there is a major public health rationale for the development of safe and effective preventive therapy (3). The technology is developing that one can consider trials for the prevention of type 1 diabetes at almost all stages of the disorder from genetic susceptibility (e.g. ongoing dietary trials such as TRIGR) to immunosuppression in patients with long-standing diabetes who receive an islet transplant(4). Trials at the onset of type 1 diabetes are the most common, and here the goal is preservation of remaining islet Beta cells (insulin secreting cells of the pancreas), which is reflected by preservation of C-peptide secretion (see below)(5).
Issues for strategies to prevent type 1 diabetes are the anticipated duration of the prophylaxis, the effectiveness of prophylaxis, the toxicity of treatment, and the complexity of the treatment regimen. If, as seems likely in man, the propensity to develop type 1A diabetes (immune mediated insulin-dependent diabetes mellitus (IDDM)) is lifelong, then young people at risk for the disease will have to be treated for many decades (unless tolerance can be restored with a time limited course of therapy). If disease susceptibility, on the other hand, depends on specific environmental factors, then prophylaxis might be tailored to match these circumstances. The Diabetes Control and Complications Trial confirmed longstanding evidence that the complications of diabetes are delayed by good control. This means that the benefits, inconveniences, and toxicity of diabetes prophylaxis must be weighed against the potential for good control, with introduction of continuous glucose monitoring(6), and especially research into monitors that will control pump insulin administration likely to change over the near term the risk benefit equation. These are likely to be related issues, because adherence to a prophylactic regimen may well require the same degree of commitment (or compliance) as does maintaining a good glycosylated hemoglobin level using existing treatments for a person with diabetes. If this is the case, then the majority of patients may fail to benefit from a prophylaxis approach, just as they fail to achieve good control with the available treatments, unless the prophylactic therapy can be administered for a limited time and have life-long efficacy. Faced with this dilemma, a rational approach would be to determine how effectively diabetes could be prevented. There are no secure data in humans yet, while there is a superfluity of data from animal models (7). The problem with these results, which are reviewed in the next section, is the ease with which it is possible to prevent diabetes in non-obese diabetic (NOD) mice and BioBreeding (BB) rats. The finding that many partially effective interventions that work in rodents have not to date worked in man must be borne in mind(8-10), and it is likely that both surrogate markers of therapeutic efficacy (e.g. assays of pathogenic T cells), more robust animal models (e.g. with 100% of animals developing diabetes), and more robust therapies (e.g. prevention not only of diabetes of but of insulitis) will be needed.
Animal Models for Type 1 Diabetes Prevention
In both BB rats and NOD mice, pancreatic beta cells are destroyed by immunological mechanisms. The strength of these models for human diabetes are discussed in Chapter 3 and extend to the characterization of several genetic predisposing factors and to the identification of pathogenic T cell clones. The weaknesses of the animal models stem mainly from our ignorance of the mechanisms by which autoimmunity is triggered or sustained in the experimental animals or, for that matter, in man. Both animal strains have been bred to maximize their incidence of diabetes and, in addition, most laboratories house their animals under conditions that yield the highest number of diabetics. Nevertheless, some 20% of female NOD mice escape disease even under "optimal" diabetogenic conditions. Whether the environmental or stochastic factors that operate to protect these mice have parallels in humans is unknown. This limits somewhat the conclusions that can be drawn from the diverse interventions that prevent IDDM in experimental models (11). With this caveat, the general approaches that have proved effective in animals are summarized in Table 12.1. In general, therapies are most effective when administered early (e.g. prior to insulitis, four weeks of age in the NOD mouse) while at onset of diabetes, fewer therapies are effective in reversing hyperglycemia. In addition most therapies, though effective in increasing the percentage of NOD animals that do not develop diabetes, fail to prevent insulitis. Insulitis persists or recurs after treatment. Animals in this condition can be unstable so that, for instance, a single injection of cyclophosphamide rapidly induces diabetes in most protected animals (perhaps by eliminating regulatory T cells). It is likely that patients who develop type 1 diabetes, who are obviously not inbred, as are the animal models, are not so finely balanced in their progression to diabetes, and more robust therapies will be required. Animal models can also be made more robust so that minimal therapies (e.g. simply injecting Freund's adjuvant) do not prevent diabetes. One example is NOD mice with the insulin 2 gene deleted. These mice more rapidly develop diabetes compared to standard NOD mice, with 100% of female mice developing disease between 7 and 15 weeks, rather than 80% between 16 and 40 weeks (12;13). At this time, it is hypothesized that therapies that either eliminate insulitis completely or are effective in more robust animal models will better predict success in man.

A recent review, generated as the company Entelos was creating a computer mathematical model of the NOD mouse suggests that careful attention to details of protocols explains much of the variance of results in the NOD mouse and such details will be important for the design of therapies in man extrapolating from the studies in the NOD (9). This review also illustrates that of 463 agents tested (321 publications) in NOD mice many failed to prevent diabetes and as an important a subset exacerbated disease including IL-12 (14), TNF alpha depended on timing of administration (15), CTLA-4 (16), PDL-1 (16), and some antigens including GAD peptides (17) and IA-2 (18). In addition, an increasing number of therapies are effective in reversing hyperglycemia of new onset NOD mice in addition to anti-CD3 antibodies (19-23) including: antilymphocyte serum plus Exendin (24), T regulatory cells CD4+ CD25+ (25), proinsulin DNA(26), complete Freund's adjuvant plus splenocytes (27) and epidermal growth factor plus gastrin (28).
Clinical Trials for the Prevention of Type 1 Diabetes
The recognition of insulin-dependent (type 1A) diabetes mellitus as an immunological disease led to the first controlled trials of interventions using nonspecific immunosuppressive treatments in subjects already diagnosed with diabetes. Cyclosporine A (CyA) and prednisone-azathioprine were tried initially because they had been effective in animals. These early trials were important because they showed that progressive loss of insulin secretion (measured with C-peptide assays) could be delayed for a period of time while immunosuppression was maintained. They also showed that the natural progression of diabetes was reinstituted following withdrawal of immunosuppression.
Trials of prophylaxis in individuals with recent-onset diabetes are based on the view that beta cell destruction is a gradual process and that an intercurrent stress precipitates symptoms (and loss of glucose homeostasis) before all insulin-making potential is lost. Unfortunately, 80 to 90 percent of islets have already been destroyed by the time diabetes is diagnosed (29). Thus, the best possible outcome of trials in people who already have diabetes is to prolong the period when small amounts of insulin continue to be made, rather than complete reversal of diabetes. Benefit to the subject under these circumstances might come not from discontinuing insulin shots but from simplifying daily blood glucose control, lowering glycosylated hemoglobin levels, and reducing the risk of hypoglycemia and diabetes complications. Benefit of this type is probable, but is unlikely to appeal to recent-onset diabetics unless the preventive regimen is simple, safe and or long-lasting with a single course of therapy. At present, measurements of C peptide are the most reliable test of endogenous insulin secretion by -cells in individuals receiving insulin injections(5). C-peptide is the connecting peptide of the proinsulin molecule that is cleaved when insulin is produced in the secretory granule. Because insulin utilized for treatment of diabetes does not contain C-peptide, C-peptide is a measure of endogenous insulin secretion. Both fasting and stimulated C-peptide is usually measured to assess the outcome of diabetes prevention trials. Given enough time, most patients (but not all) lose all of their C-peptide. Loss may occur over a number of years, and the rate of loss is usually much more rapid in children compared to adults with type 1A diabetes. Adults with type 1A diabetes who present as type 2 diabetic patients but express anti-islet autoantibodies probably have a loss of C-peptide that is so slow that practical trials (e.g. significant loss of C-peptide measured in less than three years) might be difficult, but detailed anlysis of large trials in this population is not yet available. Usually when C-peptide is preserved, metabolic control is much improved despite use of less insulin. In particular Hemoglobin A1c (measure of mean glucose over approximately a three- month time period) is reduced, and hypoglycemia may occur less frequently. It will be important to have data from long term follow up of patients with preserved C-peptide with continuous glucose monitoring to determine parameters to power trials to decreasing hypoglycemia as a potential secondary endpoint. At present, it is not clearly defined which criteria regulatory agencies will require for approval of new therapeutics. Most investigators recommend preservation of C-peptide as the primary endpoint in new-onset trials coupled with secondary endpoints of improved HbA1c and decreased variance of glucose and in particular decreased hypoglycemia.

A different approach for trials is to enter individuals identified as prediabetic by the criteria described below. The best possible outcome in prediabetic individuals (whose glucose homeostasis is not so impaired so as to make them hyperglycemic) would be the maintenance of insulin production and a life free of insulin replacement. Ascertainment of individuals with a high likelihood of developing diabetes is currently possible for relatives of patients with type 1 diabetes as well as the general population. The yield of prediabetics, about 6% of those relatives screened expressing a "biochemical" anti-islet autoantibody, is small and the prevention trials completed to date such as DPT (Diabetes Prevention Trial) have screened more than 100,000 relatives of patients with type 1 diabetes. Prevention trials in recent-onset cases are therefore likely to continue to be important, as trials with at risk individuals are very labor intensive(30).
Trials in Newly Diagnosed Subjects
Cyclosporine A (CyA)
One method of prolonging insulin production that has been successful to date is the administration of CyA (31;32). The effects of this agent were greatest when started within six weeks of initiation of insulin therapy, and the treatment had to be continued for at least six months and, preferably, for one year to show benefit. Unfortunately, the effects of CyA on T cells are lost when the CyA is discontinued. As CyA is nephrotoxic, it has been concluded, "that additional studies with CyA alone, in subjects with newly diagnosed IDDM, are no longer
Table 12.1 Examples of Interventions Preventing Type 1 Diabetes in Animal Models
Strategy |
Specificity |
Animal |
Conclusion |
Immune |
None |
NOD and BB |
Nonspecific suppression of |
Anti_CD20 |
B cells, not plasma |
NOD |
Prevention; ? Regulatory B/ Antigen presentation |
Anti-CD4 |
CD4 T Cells |
NOD |
Anti-CD4 monoclonals prevent |
Anti-CD3 |
CD3 T Cells |
NOD |
Anti-CD3 monoclonals reverse |
IL-15 Antagonist + |
Activated T Cells |
NOD |
Tolerance Induction |
Transplantation |
None |
NOD and BB |
Grafts of marrow, dendritic |
Immune Stimulation |
None |
NOD |
Immune activation by agents |
Immunologic |
Insulin/GAD/HSP60 |
NOD |
Multiple mechanisms, potential |
Oral Tolerance |
Insulin |
NOD |
Immune deviation delays |
Diets |
Unknown |
NOD |
Radical scavenging, nutritional |
Galactosylceramide |
NK T cells |
NOD |
Activation CD1 restricted cells |
Cytokines |
IL-10, TNF, IL-4 |
NOD |
Complex effect dependent |
Gene Therapy |
Antigens/Cytokines |
NOD |
Multiple potential targets |
Exenatide |
Beta Cell |
NOD |
Beta Cell Proliferation/Inhibition apoptosis |
NOD = nonobese diabetic mouse
BB = BioBreeding rat
warranted (32)." Despite this rather negative conclusion, the impact of the trials with CyA should not be underestimated. The initial promise of immunosuppression as a means to prevent diabetes stimulated interest in the early diagnosis of diabetes and led, ultimately, to screening approaches by which prediabetes could be recognized. The fact that some participants in the trials could maintain glycosylated hemoglobin (HbA1) levels in the normal range for months while on CyA showed for the first time that the destruction of beta cells could be slowed even if not halted (33). A major limitation of CyA therapy in new-onset diabetic patients is the relapse to hyperglycemia within three years even if CyA was continued. Relapses occurred despite the maintenance of C-peptide secretion. This suggested the relapses resulted from insufficient beta cell mass at the initiation of therapy rather than ineffective immunosuppression (34). The trials also stimulated awareness of the cost/benefit ratio of interventions to prevent diabetes. The costs of CyA were high, as the drug is expensive, and even with careful and expensive monitoring of blood levels, renal damage still occurred. Thus the risks were felt to outweigh the benefits.
Other Immunosuppressive Agents (Rapamycin, Mycophenolate Mofetil, Anti-IL-2 Antibodies, Methotrexate)
Using an immunosuppressive regimen consisting of Anti-IL-2R monoclonal antibodies, rapamycin (sirolimus) and low dose FK506 (tacrolimus, a calcineurin inhibitor similar to cyclosporine A), Shapiro and coworkers achieved insulin-free status in patients receiving islet transplants for type 1 diabetes (35;36). Of approximately 30 patients transplanted approximately 80% were off insulin at one year with one or more transplants. For the great majority of individuals, diabetes has recurred, a few with development of increasing anti-islet autoantibodies, but approximately half without such autoantibodies (37). Most now require low dose insulin therapy.
Mycophenolate mofetil (Cellcept) (38), an inhibitor of the synthesis of the purine guanosine monophosphate (GMP) from inosine, prevents diabetes in BB rats (39). It appears to be more potent in comparison to azathioprine, and when used with calcineurin inhibitors, has had a major benefit allowing reduced steroid dosage for pancreas transplantation.
Not yet published trial of mycophenolate mofetil alone and in combination with anti-IL2 receptor antibody in patients with new-onset diabetes (Trialnet, Peter Gottlieb) did not influence loss of C-peptide.
In a very small trial, methotrexate did not preserve C-peptide secretion in new onset patients (40).
Anti-CD3 Therapy
In the NOD mouse, monoclonal antibodies directed at the T cell receptor CD3 molecule are able to reverse hyperglycemia in acutely diabetic mice, but the same antibodies do not prevent diabetes when administered earlier in the course of disease (19;20) unless administered to neonatal NOD mice (41). There is extensive data that the anti-CD3 therapy restores tolerance and induces regulation in NOD mice(42-45).
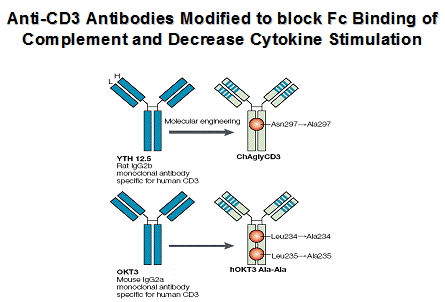
Multiple human trials are completed and phase 3 trials are underway in patients with recent onset diabetes using engineered anti-CD3 monoclonals ("humanized" and engineered to "abrogate" complement Fc binding). In contrast to standard anti-CD3 monoclonal antibodies, these antibodies do not induce severe cytokine release syndromes but have been associated with fever, rash, and in some patients adenopathy depending upon dose. Several important studies(46-48) indicate preservation of C-peptide secretion for one to two years and limited toxicity with a subset of patients developing anti-idiotypic antibodies (23). In general after 12 months it appears that progressive loss of C-peptide secretion recurs, though effects persist with a single course of therapy for up to 5 years(47). A concern is the reaction of EBV infection in the European studies, but such reactivation was self-limited with a single course of therapy (21). It is hypothesized (with good animal data re induction of T-cells expression TGF beta) and with limited human data that the larger term effects of anti-CD3 relate to induction of regulatory T-cells (42;44;49;50) including potential CD8 regulatory T cells(43;50-52). It is likely that this form of therapy will require repeated administration of the monoclonal antibody and trials are planned with repeated courses of therapy in new onset patients. Combination therapy, particularly with antigen therapy is an important consideration(53).

Insulin Metabolic Therapy
Infusion of large quantities of exogenous insulin to control blood glucose levels tightly was reported to preserve C-peptide secretion post development of diabetes (54). The stress of keeping the patient constantly in bed for 14 days with a large needle in place would probably have prevented widespread acceptance of this treatment, and there has been no replication of these studies in the subsequent two decades, and Trialnet study of parenteral insulin for diabetes prevention using annual intravenous insulin and daily modest dose insulin did not delay progression to diabetes. The Diabetes Care and Complications Trial (DCCT) has clearly demonstrated that intensive insulin therapy preserves C-peptide secretion (55). Of note a recent randomized trial in LADA patients (GAD antibody positive patients with clinical Type 2 diabetes) of insulin therapy versus oral hypoglycemic agent gliclazide demonstrated greater preservation of C-peptide with insulin therapy(56). This suggests that either insulin therapy helps to preserve beta cells or sulfonylurea therapy is deleterious.
Immunostimulation
A third approach to prolonging insulin production in the newly diagnosed diabetic is through immunostimulation (57). This strategy is based on the lower incidence of diabetes in NOD mice stimulated immunologically with agents as diverse as allogeneic
cells or the mycobacterial vaccine, bacillus Calmette-Guérin (BCG) (58). The mechanism for the protection is not clear, though stimulation and expansion of populations of T cells producing IL4 or IL10 has been suggested (58). In the absence of clear evidence linking BCG antigens with those expressed by islet cells, the mechanism of action may well be immunologically nonspecific. It was possible that immune stimulation after the onset of diabetes might reduce insulin requirements or increase C-peptide synthesis when measured one year after vaccination with the attenuated mycobacterium, BCG. Vaccination with BCG is thought to be safe and is immunogenic, and a preliminary small human study gave some evidence for benefit in preserving C-peptide. We have completed a double-blind study (59) employing two medical teams, one to manage the diabetes and one to administer and read the BCG skin response and placebo vaccines. Entry was restricted to subjects within eight weeks of clinical onset, or within 16 weeks if the islet cell antibody (ICA) test is positive, who are between ages 5 and 18 years and who have negative human immunodeficiency virus (HIV) and purified protein derivative (tuberculin) (PPD) tests. BCG vaccination did not preserve C-peptide secretion, as found in other studies (59). A report by Bonifacio and co-workers indicates that neonatal BCG vaccination did not influence the development of islet autoantibodies in a prospectively followed cohort of offspring of patients with type 1diabetes (4.9% and 5.2% for development of multiple autoantibodies), but progression to diabetes was significantly faster for the autoantibody positive children who were vaccinated (74% vs. 37%; p=.05) (60). In a similar manner, Q-fever vaccine apparently had no effect in new onset patients (Harrison, oral communication).
Treatments to halt islet cell destruction and prevent diabetes would ideally be started before the beta cell mass is significantly compromised. Nevertheless, detecting partial benefit in recent-onset diabetics would be useful if it is a precursor to an intervention that could be used with greater efficiency in subjects with pre-type 1 diabetes. Ultimately, the focus of the prevention of diabetes must be in patients with pre-type 1 diabetes (hereafter referred to as prediabetes), and the remainder of this chapter reviews progress toward this goal.
Identification of Subjects with Pre-Type 1 Diabetes
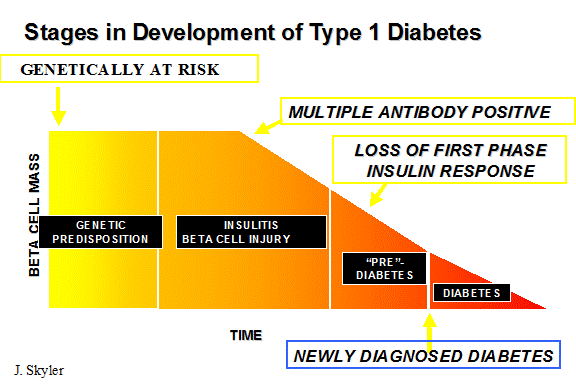
Prediabetes describes the period when islet cell destruction is in progress but glucose homeostasis has not been lost. The best definition of a prediabetic is an individual who actually develops diabetes. Subjects are currently identified as being at high risk by the sequence of events summarized below:
1. They are ascertained by their relationship to a diabetic individual (usually a first-degree relative) or identified to have high-risk HLA genotypes such as DR3-DQ2/DR4-DQ8.
2. Their serum has tested positive for immunological markers of islet specific immunity (the presence of antibodies associated with islet beta cells).
3. Their first phase insulin production (see Metabolic Testing, below) is low enough to give at least a 50% or greater risk for diabetes within the next five years and/or they have impaired fasting glucose or impaired glucose tolerance (e.g. two hour glucose between 126 and 200 mg%).
These selection criteria in effect reproduce two of the factors thought to be important in the etiology of type 1 diabetes: genetic predisposition and the development of autoimmunity.
Genetics
MHC Genes. When one pair of identical twins develops type 1 diabetes, the second twin has a greater than 50% risk of also developing diabetes (see Chapter 7)(61). The findings that fewer than 100% of second twins become diabetic suggest that additional factors, probably environmental, contribute to the pathogenesis of type 1 diabetes. Similarly, a higher risk for diabetes was shown for siblings (approximately a 5-10% risk) or for offspring when the father had diabetes (approximately 12% risk) or when the mother had diabetes (approximately a 6% risk) (62). The strongest genetic associations are with the human leukocyte antigen (HLA) system. Approximately 95% of people with type 1 diabetes have either DR3 or DR4 (compared to 50% of the general population), and 30 to 53% have the DR3 and DR4 combination (compared to 2.4% of the general population in Denver, Colorado) (63). A subset of individuals at extreme risk for Type 1 diabetes can be identified such as DR3/4-DQ2/8 siblings of patients with type 1 diabetes who have inherited both HLA haplotypes identical by descent with their proband sibling. Our risk estimates for development of islet autoantibodies, followed by diabetes is as high as 80%(64) for such siblings. Adding analysis of DP alleles to DR and DQ typing risks in general population for DR3/4 heterozygous individuals are as high as 20%(65).
A combination of assays for anti-islet autoantibodies (GAD65, ICA512, insulin, ZnT8) can identify high-risk individuals(66). Usually the presence of multiple anti-islet autoantibodies is associated with at-risk HLA alleles(67). Relatives of patients expressing two or more autoantibodies have a greater than 50% five- year risk, with most progressing to diabetes within ten years.
Much of the contribution from HLA DR4 stems from linkage with an HLA DQ allele DQ8 (DQB1*0302), with an influence of DR4 subtypes (e.g. DRB1*0403 decreases risk even on DQB1*0302 containing haplotypes). The allele DQB1*0302 lacks aspartic acid at position 57 of the DQB chain (Asp-negativity), and a lack of aspartic acid is associated with high risk for only a subset of DQ alleles (68;69). In Finland, Asp-negativity was found in 74.5% of patients with type 1 diabetes compared to 18% of controls, but for the most part, this simply reflects the presence of the highest risk HLA DQ alleles DQ2 and DQ8 (DQB1*0302) (70). It is important to realize that absence of asp57 is only an approximate rule, and that alleles such as DQB1*0401 that provide high risk have asp at position 57, so typing for full DQ allele sequences is important (25). Several HLA alleles are associated with a very low incidence of diabetes (see Chapter 7). Thus, genetic screening is likely to play an important role in identifying subjects who could be enrolled in prevention trials. The present generation of trials will enroll high-risk individuals identified among first-degree relatives of index cases. Because 70-90% of patients have no first-degree relative with diabetes (71), this creates a potential for significant ascertainment bias. For example, first-degree relatives of patients with diabetes presenting in infancy are likely to be underrepresented merely because family sizes are small and parents may be discouraged from having more children where the first has a time-consuming and emotionally demanding condition ("stoppage"). First-degree relatives of parents who develop diabetes after 40 years of age may be over represented merely because family size will not have been limited in response to parental disease. How important these sources of bias are will depend on the diversity of genes contributing to disease susceptibility.
Studies are underway following HLA typed infants from birth. In the Denver Colorado (DAISY study), 2.4% of newborns have the genotype DR3/4, DQ2/8(DQ8=DQB1*0302). Approximately 50% of children presenting with diabetes before age five have this genotype. It is estimated that children from the general population (no relative with type 1 diabetes) with DR3/4, DQ2/8 have a risk of diabetes of approximately 1/16, similar to the overall risk of first-degree relatives. In contrast, a sibling of a patient with type 1 diabetes with DQ2/DQ8 has a risk of activating autoimmunity (e.g. islet cell autoantibodies) of approximately 50%. Though high risk can now be identified even in the general population with antibody testing, HLA evaluation has a relatively low sensitivity compared to conventional newborn screening programs. Studies however can be designed to evaluate novel therapies, even in newborns (e.g. Finnish studies currently underway, DIPP (72)) and if a safe and effective therapy is documented, it would likely find wide use.
Non-MHC Genes. Animal studies indicate multiple genes contribute to susceptibility to diabetes. An advantage to identifying pre-diabetics by screening first-degree relatives is that selection for non-HLA genes is likely to occur in parallel (73). The development of multiple markers for polymorphisms on each human chromosome is making possible the identification of non-MHC genes predisposing to diabetes, so that screening of the general population will be more productive in the long-term. At present, polymorphisms of more than 40 genetic loci are associated with Type 1 diabetes(74) including HLA, the insulin gene, a lymphocyte tyrosine phosphatase (LYP, PTPN22), IFIh1, IL2 receptor and CTLA-4.
As more genetic markers associated with susceptibility and protection are identified, it will be important to apply them retrospectively to the prevention protocols where high-risk people are initially identified with immunological tests. For example, if immunological and metabolic tests indicate a high risk for diabetes and the subject is entered into a prevention protocol, it will be important to make sure that relevant genes are similarly divided in numbers of subjects with high risk and lack of protective HLA alleles. Cells might also be saved for other markers that may be shown to be good predictors in future years, as this area of research is advancing rapidly.
Immunological
Some reports have suggested that the degree of ICA positivity is the best predictor for advancement to insulin dependence (75-77). It is likely that the ICA test will be replaced by "biochemical autoantibody" determination (67;78).
Others consider ICA positivity only to be an indicator that damage has occurred to the islet, and insist that a metabolic test (see below) be done to determine the amount of damage. We previously noted that nine of ten first-degree relatives advancing to diabetes had ICA titers greater than or equal to 80 JDF units, making this a high-risk finding (79). However, six other first-degree relatives of diabetics had similar titers of ICA and had not advanced to diabetes over a six-year period. Thus, the high titer results might have a relatively poor positive predictive value (10 of 16 subjects). Also, subjects with lower titers of ICA obviously also develop diabetes, although the percentages are not as high. Bruining and co-workers (80) noted that only four of eight children found to have positive ICAs in a general population (not first-degree relatives) developed diabetes in a ten-year period. Another 3 of 4806 children initially negative for ICA also developed diabetes. Thus, whether screening first-degree relatives or the general population, it would seem that a second test, more specific for quantity of damage, would be important and the DPT-1 (Diabetes Prevention Trial Type 1) used such a structure (81). It is now clear that a combination of immunologic tests (e.g. multiple anti-islet autoantibodies(82), high affinity insulin autoantibodies(83)) combined with metabolic evaluation can improve prediction of risk and time to diabetes.
ICA assays are difficult to quantitate and this leads to interlaboratory differences. Factors that are likely to limit the reproducibility of the assay between laboratories include (1) quality of the human pancreas chosen for the assay and (2) interobserver differences for a subjective endpoint. Nevertheless, the Third International Workshop on ICA concluded, "The indirect immunofluorescent of (IFL) test for ICA is still the most useful screen for identifying people at risk for type 1 diabetes, and, until such time as the relevant antigens are identified in purified in large quantities, it remains the method of choice for its detection (84)". Multiple autoantibodies have been identified in the blood of newly diagnosed subjects (and prediabetic subjects). These include autoantibodies to insulin (IAA) (85;86), glutamic acid decarboxylase (GAD) (87-89), and IA-2 [ICA512] (90). With the three islet autoantigens that have been isolated, there are standard radioassays; namely assays for autoantibodies reacting with insulin, with glutamic acid decarboxylase, and with the molecule ICA512 (IA-2). These radioassays are typically performed in 96-well filtration plates and are also commercially available. A high-throughput laboratory can probably perform each assay for a cost less than $20 per sample and thus large scale screening just using the biochemical assays is feasible. A modified ELISA-like assay (Kronus) is available for GAD65 autoantibodies that performs well in international standardization studies, though in the 2009 workshop with excellent sensitivity, specificity for many laboratories utilizing this assay was only 95%(Mueller oral report), while many of the best radioassays had higher specificities (with slightly lower sensitivities). This may relate simply to the threshold setting for the assay.
The presence of two or more of the four autoantibodies (insulin, GAD65, ICA512(IA-2), ZnT8) indicates a high risk of progression to type 1 diabetes (see chapter on humoral autoimmunity). The risk exceeds 50% in five years even without the additional information provided by intravenous glucose tolerance testing. Nevertheless, for any combination of autoantibodies, loss of first phase insulin secretion decreases the time to diabetes. Of note, an individual with a single anti-islet autoantibody (overall risk approximately 20%) who has first phase insulin secretion below the first percentile has a diabetes risk exceeding 50% in five years. Another high-risk feature is the presence of impaired fasting or impaired glucose tolerance on oral glucose tolerance testing (e.g. two- hour glucose between 126 and 200 mg%).
Metabolic testing
The interval between detecting an autoantibody in a first-degree relative of a diabetic and the development of diabetes can exceed ten years or be as short as months. Prospective studies where the success of an intervention will not be known for more than ten years are clearly not practical, so a further selection for subjects with extensive beta cell destruction is usual. Direct measurement of the functional mass of islet cells is not possible; the best approximation comes from the intravenous glucose tolerance test (IVGTT) and assessment of impaired fasting and glucose tolerance. Srikanta and coworkers (91) were the first to show that the sum of the 1- and 3-minute insulin levels had some predictive value. Experience from the Barbara Davis Center confirmed the predictive value of a very low first-phase insulin response (FPIR) on IVGTT (<25U/ml) in children (92). A later report (17) noted predictability for different levels of FPIR in first-degree relatives under age 18 years who had two or more positive ICA tests (Table 12.2). Vardi and coworkers (44) found 15 of 17 (88%) ICA-positive first-degree relatives develop insulin dependence if their FPIR fell to <48 U/ml (the first percentile at the Joslin Diabetes Center). In contrast, only 3 of 18 (17%) with values above this level developed insulin dependence. Low first phase insulin secretion has been documented in anti-islet autoantibody positive children less than age five at their first testing(93). This is probably related to their risk of very rapid progression to diabetes.
Little is known about the replicate errors, either within subject or between subjects, of the IVGTT. The test needs to be performed with consistency and utmost accuracy in timing (94;95). It should be performed after an overnight fast of uniform duration. Some people vary in their response, and the downward progression of the FPIR is not always linear (92). Finally, the results are age-dependent, particularly in prepubertal versus pubertal children (96).
The simplest assay for helping to define metabolic progression to Type 1 diabetes is the HbA1c assay. Over the last one to two years prior to onset of diabetes amongst the great majority of islet autoantibody positive individuals there is a progressive rise of HbA1c in the normal range(97).
Trials Using Subjects with Pre-Type 1 Diabetes
An NIH consensus report on prevention of type 1 diabetes concluded: "The general consensus from the meeting was that indeed there is methodology that can identify, with near certainty among first-degree relatives of type 1 diabetic patients, those who will develop diabetes and that immune intervention therapy before the onset of symptoms might prevent the disease from occurring (98)."
Table 12.2. FPIR* and Time to Diabetes (79)
Time to diabetes (months)
FPIR* (mU/ml) Mean time 95th percentile
<67 19 57
<46 14 21
<30 4.6 18
*FPIR = First phase insulin response (sum of 1-minute 3-minute insulin levels) on intravenous glucose tolerance testing.
It is now generally accepted that prediabetes can be accurately diagnosed in select high-risk groups of subjects. The justification for identifying this high-risk group comes from their potential recruitment into protocols to test strategies to prevent diabetes as well as avoiding morbidity and the risk of death at onset (different countries have reported rates between 1/200 and 1/500)(99;100). These high-risk subjects are, however, so rare that no center is likely to identify more than 20 subjects a year who would be eligible to enter a prevention trial. Identifying prediabetics is very expensive (we estimate $1,200 per subject, allowing for autoantibody screening and two IVGTT tests), so a cost-effective approach requires extensive and international collaboration.
The largest prediabetic prevention studies to date have evaluated the effects of insulin (parenteral, nasal and oral administration) and nicotinamide(101-106). Smaller studies have evaluated other agents.
Table 12.3 Prevention Type 1A Diabetes Onset
Trial |
Agent |
Comment |
TRIGR |
Casein Hydrolysate |
Genetically Susceptible (Pilot approx |
DIPP (DM Prediction/ |
Nasal Insulin |
No Effect Post Appearance first anti-islet |
DPT-1 (DM prevention Trial) |
Parenteral Insulin |
No Effect |
|
Oral Insulin |
No Effect/Potential + subgroup high insulin autoantibodies |
DENIS (Deutsche |
Nicotinamide |
No Effect |
ENDIT (European |
Nicotinamide |
No Effect |
Melbourne Nasal Insulin |
Nasal Insulin |
Crossover-Immune Monitoring – |
Carel Cyclosporine |
Cyclosporine |
No Permanent Prevention |
Bohmer Ketotifen |
Histamine Antagonist |
No Effect |
Hummel Gluten |
Gluten Elimination |
No Effect |
Nicotinamide
The mechanism of nicotinamide protection is currently unknown (107). Nicotinamide was shown initially to delay diabetes in NOD mice (108). Nicotinamide inhibits poly(ADP-ribose) synthetase (109) and, at high concentrations, can act as a free radical scavenger (110). Pre-treatment with nicotinamide prevents the decrease in proinsulin synthesis in islets from rats treated with alloxan or streptozotocin (109). It also inhibits the activated macrophage killing of beta cells in vitro (111)and the expression of class 2 major histocompatibility complex (MHC) on beta cells caused by cytokines, tumor necrosis factor (TNF), and interferon gamma (112). All these processes are thought to be part of the immune-mediated destruction of beta cells found in the human disease. A workshop report summarizes the biological action and therapeutic potential of nicotinamide (113). A number of studies have evaluated nicotinamide in new-onset patients with reports that some preservation of C-peptide may occur, but only in older individuals (114). In well-controlled trials in children, there was no effect (115).
A pilot study of nicotinamide was undertaken jointly between Auckland, NZ, and Denver, U.S.A. (116). Among 14 treated subjects, only one developed diabetes after a mean of 17 months of treatment, whereas eight untreated subjects were all insulin-dependent after a mean of 17 months. All subjects were less than 16 years old when diabetes was detected by ICA screening and all had ICA titers equal to or greater than 80 JDF units. All subjects had a FPIR of less than 67 U/ml (the 5th percentile of normal). On further (unpublished) follow-up of approximately five years, three of four subjects in Denver (all of who had a FPIR <30 U/ml) and five of ten Auckland subjects (most with the higher FPIR levels) had become insulin dependent. This pilot trial was weakened by the fact that all the control subjects were followed in Denver.
Nicotinamide was evaluated in a German prevention trial headed by Dr. Hubert Kolb (Diabetes-Forschunginstitut an der Universitat Dusseldorf) and in the European Nicotinamide Diabetes Intervention Trial (ENDIT) study under the direction of Dr. Edwin Gale (Department of Endocrinology and Diabetes at the University of Bristol). The German trial was named "DENIS" for Deutsche Nicotinamide Intervention Study. The DENIS trial was ended without detecting any difference in progression to diabetes between the placebo and nicotinamide treated groups (59). The trial was relatively small and met criteria for early discontinuation.
The ENDIT trial was a double blind, randomized, and placebo-controlled trial (117). Enrollment was open to first-degree relatives of patients with type 1 diabetes (sibling, child, parent) who had at least two ICA-positive tests drawn 3-12 months apart, one equal to or greater than 20 JDF units.. An IVGTT was performed by the ICARUS protocol (95) and was used for the subsequent meta-analysis but not for randomization. An oral glucose tolerance test (OGTT) was performed as a two-point test and (0 min and 2 h capillary blood glucose) to rule out diabetes mellitus by World Health Organization (WHO) criteria (118). Demographic information was collected following the example of the ICARUS questionnaire. Follow-up procedures include yearly FPIR with ICA and IAA tests(119). The dose of nicotinamide was 1.2 g/M2 per day.
The primary outcome measure was the incidence of diabetes mellitus by WHO criteria. The OGTT was performed at least once per year. Nicotinamide did not slow progression to diabetes (103). In addition only approximately half of relatives with cytoplasmic ICA autoantibodies had biochemical autoantibodies, and there was minimal progression to diabetes for relatives only expressing cytoplasmic ICA autoantibodies(102). Thus prediction was possible, but nicotinamide was without benefit.
Insulin
The mechanism by which insulin might delay or prevent diabetes is complex. It was initially hypothesized that the beta cells were allowed to "rest" and might therefore be less prone to immunological attack. This may be the case for the BB rat model where only biologically active insulin prevents disease, and insulin must be administered in amounts that produce hypoglycemia (120;121). Additional mechanisms, such as induction of immunoregulation/"tolerance," contribute to protection in the NOD mouse model (122-128). The initial pilot study of insulin in subjects with prediabetes was reported from the Joslin Clinic (129).
The very small number of subjects treated is a weakness of this pilot trial, as was the inclusion of two subjects with FPIR levels of 76 and 83 U/ml (normal) in the treatment arm. A much larger, appropriately powered trial has evaluated IV, subcutaneous, and oral insulin (for induction of oral tolerance) with a multicenter trial under the auspices of the National Institutes of Health (NIH). The criteria for randomization into the parenteral DPT-1 trial are summarized in Table 12.4.
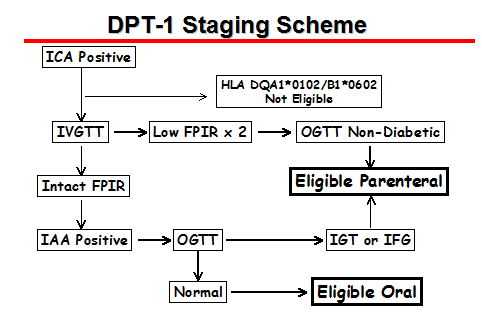
For this DPT-1 trial (Diabetes Prevention Trial Type 1), more than 100,000 first-degree relatives of patients with type 1 diabetes have been screened for the expression of cytoplasmic islet cell antibodies (ICA). There were two separate trials in DPT-1: parenteral insulin administration and a trial of oral insulin. Approximately 4% of the screened subjects were ICA positive. One half of the ICA positive individuals, however, did not express either GAD65 or ICA512 (IA-2) autoantibodies, and these individuals had only rarely lost the FPIR that would make them eligible for the parenteral insulin intervention phase of DPT-1 (78). The parenteral insulin prophylaxis study randomized 339 subjects to either close observation (no therapy) or intravenous insulin for four days once a year and 0.125 U/kg body weight of subcutaneous ultralente insulin twice daily. Analysis of progression to diabetes (as defined by oral glucose tolerance testing) indicated that the insulin therapy did not delay progression to diabetes (81). The DPT-1 trial was designed in the early 90's. It has subsequently become apparent that in animal models, such as the NOD mouse, a major effect of insulin appears to relate to induction of immunoregulatory T cells; the dose chosen in man (necessary for safety to minimize the risk of hypoglycemia) is ineffective in the NOD model and thus if a higher dose is considered the insulin must be metabolically inactive (130). Despite the lack of therapeutic benefit, the completed parenteral DPT-1 trial is providing a wealth of information concerning prediction of type 1A diabetes and trial design. It is a testament to the seriousness of the disorder that so many individuals were screened and willing to participate for years in such an intensive trial regimen. It appears that future trials will be greatly streamlined by screening with "biochemical autoantibody" assays. Almost 1/20 of the relatives with GAD or ICA512 autoantibodies do not have cytoplasmic ICA, and the ICA positive relatives lacking GAD and ICA512 autoantibodies appear to have a low risk of progression to diabetes (78). The presence of multiple biochemical anti-islet autoantibodies (of GAD65, ICA512, insulin or ZnT8) is likely (but further analysis is necessary) to provide enough prognostic information that even the intravenous glucose tolerance test may be optional for assigning risk values (67).
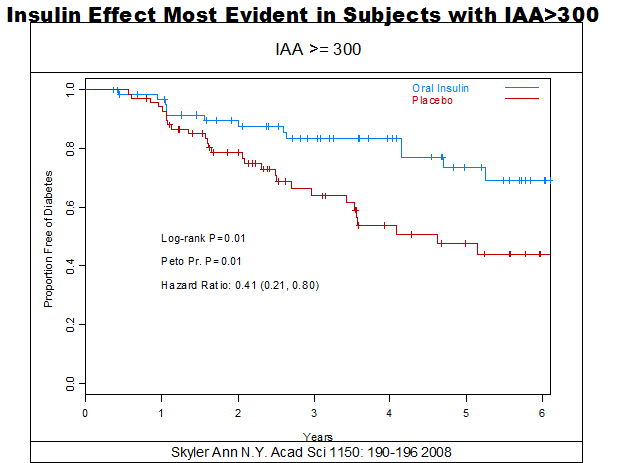
The second trial within DPT-1, namely the oral insulin trial, was reported at the American Diabetes Association meeting (2003) (131). Three hundred and seventy-two antibody positive relatives have entered into this randomized, double blind trial with a defined risk of diabetes of 26-80%. Ninety-seven thousand samples were initially screened to identify the group with this risk and the group entered into the parenteral insulin trial. Entry criteria are similar to those for the parenteral DPT-1 trial with the exception that first phase insulin secretion had to exceed those assigned for high risk in the parenteral trial, oral glucose tolerance cannot be impaired, and insulin autoantibodies must be present. This identifies a subgroup with a predicted risk of diabetes between 25-50% within five years. Similar to the parenteral insulin trial, oral insulin versus placebo did not overall slow progression to diabetes. In the NOD mouse model, oral insulin delayed progression to diabetes for a subset only. A substantial percentage of mice progressed to diabetes despite oral insulin, and insulitis remained. Of note, for relatives with high levels of insulin autoantibodies only, oral insulin may have delayed but did not prevent progression to diabetes. For those relatives with levels of insulin autoantibodies greater than 200nU/ml, a delay in progression to diabetes of approximately 10 years may have been associated with the therapy(4) but this is a subset analysis.
The DPT-1 trial groups formed TrialNet in October 2001 to create a North American network for conducting trials for the prevention of type 1 diabetes and international sites were added. TrialNet will evaluate therapies in new-onset patients as well as pre-diabetes and is open to suggested therapies and protocols. With such a collaborative network, it is hoped that the discovery of effective and safe therapies will be hastened. There are additional major efforts to develop therapies for diabetes prevention. In particular, the Immune Tolerance Network (www.immmunetolerance.org) is accepting applications to support therapies aimed at tolerance induction and assays of tolerance (132). The Immune Tolerance Network, sponsored by the National Institutes of Health, funds studies both within and outside of the United States.
Nasal insulin was evaluated in a novel and ambitious study from Finland (DIPP – Diabetes Prediction and Prevention (72)) where infants are followed from birth. When ICA was found, the children were randomized to either nasal insulin therapy or placebo groups. Again ability to predict and conduct an excellent trial were demonstrated but the therapy did not delay progression to diabetes(106).
Another international study (TRIGR) is evaluating exclusion of bovine milk in early childhood (133) with preliminary data. There is contradictory data relative to the epidemiologic analysis of bovine milk exposure related to diabetes risk(106;134) with a recent manuscript reporting heterogeneity related to the PTPN22 genetic polymorphism associated with diabetes risk(135). The TRIGR trial is now fully enrolled and should provide a definitive answer as to importance of early introduction of bovine milk related to etiology of Type 1 diabetes.
for High-Risk (Parenteral Insulin) Protocol
First-degree relatives: 4 to 45 years (inclusive)
Second-degree relatives: 4 to 20 years (inclusive)
ICA > 10 JDF units (2 of 3 times in series)
Not DQA1*0102, DQB1*0602
IVGTT criteria:
OGTT not diabetic (ADA Expert Committee Criteria)
ICA=Islet cell antibodies
IVGTT=Intravenous glucose tolerance test
OGTT=Oral glucose tolerance test
NDDG=National Diabetes Data Group
Additional Clinical Trials of Antigen Based Therapies
At least four islet antigens in addition to intact insulin have entered clinical trials for preservation of insulin secretion at diabetes onset: an altered peptide ligand of insulin peptide B:9-23 (Neurocrine) (136;137), the GAD65 molecule (Diamyd) (138;139), a heat shock protein peptide (Peptor) (Table 12.5) (140;141) and the HLA-DR4 restricte proinsulin peptide C19-A3(142).
A phase I/II trial to evaluate an altered peptide ligand of the immunodominant insulin B:9-23 peptide that Daniel and coworkers found to be a major target of intra-islet T cells. This peptide could, by multiple routes of administration, prevent diabetes (143-145). The peptide (mouse insulin 2 and human insulin) is identical in man and mice. Recent studies using ELISPOT assays indicate that patients with or developing type 1 diabetes also recognize this molecule (136). There are several important caveats relative to peptide immunotherapy. Even with a self-peptide, one can induce anaphylaxis; we have induced anaphylaxis in NOD mice with the native B:9-23 peptide (146). A self-peptide may activate disease, and we have induced insulin autoantibodies in normal Balb/c mice and NOD mice with the insulin B:9-23 peptide (137;147). The altered peptide ligand had no effect on loss of C-peptide in new onset trial(oral communication Anette Ziegler).
The whole GAD65 molecule has entered clinical trails in Scandinavia in patients with LADA (latent autoimmune diabetes of adults) and a completed phase II study in new onset Type 1 diabetes reported(148). This usually slower form of diabetes may be a difficult therapeutic target. Oral description of the trial at the American Diabetes Association (2003) indicated minimal loss of C-peptide in placebo patients, making demonstration of preservation of C-peptide by GAD65 immunization a difficult task. At one dose (20ug) in the phase II study there was reported significant preservation of C-petpide(149). Injections did not have side effects and in particular there is no evidence that Stiff Man Syndrome was induced. A controlled trial in new onset patients with Type 1 diabetes with a single injection of GAD65 in alum demonstrated decreased loss of C-peptide secretion(150) but without effect on either HbA1c or insulin utilization. Repeat studies and a Trailnet preventive trial are planned.
Table 12.5 Trials to Preserve Beta Cells post Diabetes Onset
Trial |
Agent |
Comment |
Gainesville Azathioprine |
Azathioprine+Glucocorticoid |
Minimal Delay Loss C-Peptide |
Canada/ Europe |
Cyclosporin A |
Preservation C-Peptide only During Administration |
Eisenbarth ATGAM |
Anti-thymocyte Globulin |
No Lasting Effect |
Buckingham Methotrexate |
Methotrexate |
No Effect |
Herold Anti-CD3 |
Ala-Ala anti-CD3 |
? No Lasting Effect with single |
Chatenoud Anti-CD3 |
Deglycosylate anti-CD3 |
Confirms up to 2 year delay loss C-peptide of Herold trial; transient EBV reactivation |
Pescovitz, Trialnet |
Anti-CD20 |
Protective effect oral presentation ADA 2009 meeting |
Denver BCG |
BCG Immunization |
No Effect |
Brod Interferon |
Oral Interferon Alpha |
Ongoing |
Australia Q Fever |
Q Fever Vaccine |
No Effect |
Diamyd GAD |
Human GAD in Alum |
Delay loss C-peptide but no effect HbA1c, insulin utilization |
Peptor p277 |
Heat Shock Protein Peptide |
No effect children, Negative small follow up study adults |
p227 LADA Patients |
|
Ongoing |
Oral Insulin Italy/France |
Oral Insulin |
No Effect |
NBI-6024 Neurocrine |
Altered Peptide Ligand Insulin |
No Effect |
TrialNet Gottlieb |
Anti-IL2 Receptor and Mycophenolate Mofetil |
No Effect |
Results of a small Israeli trial in new onset adults suggested preservation of C-peptide with relatively short follow up (151), while a follow up small study did not find any preservation in children and non-significant trend in adults(152;153). A trial in LADA patients is underway(153). A difficulty with the p277 peptide therapy is the lack of convincing rationale that HSP60 is an autoantigen in the animal model (no workshop demonstrated anti-HSP60 autoantibodies or T cell reactivity) and conflicting reports for therapeutic efficacy even in the NOD mouse (154). It has recently been suggested that HSP60 may activate the innate immune system (Toll activator) (155-157), and, surprisingly, the peptide might have a similar effect (158). Such studies are complicated by the frequent contamination of protein molecules by lipopolysaccharide (LPS) where small amounts of LPS activate Toll 4 receptors and have important in vivo effects (159).
Cellular Therapies
A number of approaches are being pursued in terms of cellular therapies to prevent beta cell destruction with initial human trials planned and underway for some of the approaches. In particular there is a large preclinical effort to utilize regulatory T cells to modulate autoimmunity as well as autologous stem cells and antigen presenting cells.
Perhaps the simplest trial was administration of autologous cord blood to patients with Type 1 diabetes, where cord blood had been frozen at birth(Schatz et al oral presentation ADA 2009;A7). With 23 patients treated and 15 with one year of at lest one year of follow up it can only be concluded in the absence of control population that C-peptide loss continues after diabetes onset in the majority of individuals.
There is a large body of evidence demonstrating the importance of regulatory T cells for the natural prevention of Type 1 diabetes. With the rare mutation of the foxP3 gene that controls development of a major subset of regulatory T cells it is estimated that 80% of children develop Type 1 diabetes in addition to multiple other autoimmune disorders. In preclinical studies regulatory T cells have been expanded and are able to prevent diabetes of the NOD mouse(160). Human regulatory T cell s can be expanded in vitro(161) though their stability as a regulatory T cell is questioned(162). Initial human trials will likely utilize mixtures of regulatory T cells, though antigen specific T cells provide greater efficacy in preclinical studies.
Another proposed cellular therapy utilizes dendritic cells, either pulsed with antigen or modulated in vitro to be toleragenic(163;164). It is likely that the initial clinical trials will utilize dendritic cells without antigen pulsing.
Conclusions
The fact that diabetes can be successfully managed and complications delayed by good control of blood glucose has placed significant constraints on treatments intended to prevent diabetes. Global immunosuppression protocols, of the types used for transplant recipients, clearly carry an unacceptable morbidity, though a trail in Brazil of cyclophosphamide combined with anti-thymocyte globulin and autologous stem cell transplantation in new onset patients has been carried out(165). Though there is evidence of delay of loss of C-peptide for a subset of patients, with 12 patients maintaining an insulin independent status with 8 relapsed the morbidity (and potential mortality) already demonstrated should rule out this therapeutic regimen in new onset patients(165). A trial of rabiit anti-thymocyte globulin without cyclophospamide is underway in the United States. A small trial of horse anti-thymocyte globulin though with reported extension of "honeymoon" phase and inversion of CD4/CD8 ratio similar to current anti-CD3 trials was stopped due to induction of thrombocytopenia in two patients(166).
Immunologists caring for patients with autoimmune diseases have a need to establish organ-specific immunological tolerance that is just as pressing as that facing transplant surgeons. The approaches that have been explored to date in large preventive trials seem very speculative when viewed in relation to the complexity of immunoregulation and the development of autoimmunity (167). The lack of surrogate markers for the desired therapeutic T cell effects complicates interpretation. Potential surrogate markers for T cells are just becoming available for the NOD mouse (168) and man(169). Until they are available, clinical studies of diabetes prevention will be as difficult as attempts to treat hypertensive kidney disease without being able to measure blood pressure. The animal models of type 1 diabetes should be a fruitful ground for treatments designed to prevent diabetes in humans. In reality, it has been difficult to realize this expectation. One obstacle has been the ease with which diabetes is prevented in NOD mice (7). For example, interventions that have succeeded in mice include special diets, injection with male sex hormones, and the failure to exclude common mouse viruses from colonies.
The limitations of animal models necessitate the development of prevention approaches in humans. Constraints on research funding on both sides of the Atlantic have increased the pressure for collaboration beyond local and national boundaries. The initiation of TrialNet and large multi-center trials (e.g. Immune Tolerance Network) to determine whether type 1 diabetes can be prevented or ameliorated at onset promises to make this decade an exciting one. Both of the above organizations welcome applications for protocols for diabetes prevention. In particular the Immune Tolerance Network has an online application process (immunetolerance.org) and funds U.S. and international trials with potential to induce tolerance. Trialnet has a similar goal for international collaboration and funding.
Reference List
(1) Mueller PW, Bingley PJ, Bonifacio E, Steinberg KK, Sampson EJ. Predicting type 1 diabetes using autoantibodies: the latest results from the diabetes autoantibody standardization program. Diabetes Technol Ther 2002;4(3):397-400.
(2) Bingley PJ, Bonifacio E, Mueller PW. Diabetes antibody standardization program: first assay proficiency evaluation. diab 2003 May;52(5):1128-36.
(3) Maniatis AK, Yu L, Miao D, Nelson K, Eisenbarth GS. Rapid Assays for Detection of Anti-islet Autoantibodies: Implications for Organ Donor Screening. J Autoimmun 2001;16(1):71-6.
(4) Skyler JS. Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci 2008 December;1150:190-6.
(5) Palmer JP. C-peptide in the natural history of type 1 diabetes. Diabetes Metab Res Rev 2009 May;25(4):325-8.
(6) Garg SK, Kelly WC, Voelmle MK, Ritchie PJ, Gottlieb PA, McFann KK, Ellis SL. Continuous home monitoring of glucose: improved glycemic control with real-life use of continuous glucose sensors in adult subjects with type 1 diabetes. Diab care 2007 December;30(12):3023-5.
(7) Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999 June;5(6):601-4.
(8) Atkinson MA. ADA Outstanding Scientific Achievement Lecture 2004. Thirty years of investigating the autoimmune basis for type 1 diabetes: why can't we prevent or reverse this disease? Diabetes 2005 May;54(5):1253-63.
(9) Shoda LK, Young DL, Ramanujan S, Whiting CC, Atkinson MA, Bluestone JA, Eisenbarth GS, Mathis D, Rossini AA, Campbell SE, Kahn R, Kreuwel HT. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005 August;23(2):115-26.
(10) Roep BO, Atkinson M. Animal models have little to teach us about type 1 diabetes: 1. In support of this proposal. diabetol 2004 October;47(10):1650-6.
(11) Haskins K, Atkinson M. Prediction of Diabetes and Response to Therapy in Individual Animals: are there Lessons for Man? Report on the JDF/Snow Mountain Meeting, 5-7 October 2000, Winter Park, CO, USA. J Autoimmun 2001 February;16(1):15-9.
(12) Thebault-Baumont K, Dubois-LaForgue D, Krief P, Briand JP, Halbout P, Vallon-Geoffroy K, Morin J, Laloux V, Lehuen A, Carel JC, Jami J, Muller S, Boitard C. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest 2003 March 15;111(6):851-7.
(13) Moriyama H, Paronen J, Liu E, Abiru N., Melanitou E., Babu S, Bao F, Sikora K, Gianani R, Eisenbarth GS. Testing the hypothesis that the insulin 1 gene is essential for diabetogenesis and knockout of insulin 2 gene accelerates diabetes in NOD mice. Diabetes/Metabolism Research and Reviews 2002;18(Suppl 4):S26.
(14) Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med 1995;181:817-21.
(15) Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes [published erratum appears in Immunity 1997 Feb;6(2):following 215]. Immunity 1996 September;5(3):285-93.
(16) Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, Auchincloss H, Jr., Sayegh MH. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 2003 July 7;198(1):63-9.
(17) Cetkovic-Cvrlje M, Gerling IC, Muir A, Atkinson MA, Elliott JF, Leiter EH. Retardation or acceleration of diabetes in NOD/Lt mice mediated by intrathymic administration of candidate b-cell antigens. diab 1997;46:1975-82.
(18) Trembleau S, Penna G, Gregori S, Magistrelli G, Isacchi A, Adorini L. Early Th1 response in unprimed nonobese diabetic mice to the tyrosine phosphatase-like insulinoma-associated protein 2, an autoantigen in type 1 diabetes. J Immunol 2000 December 15;165(12):6748-55.
(19) Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA 1994 January 4;91(1):123-7.
(20) Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 1997 March 15;158(6):2947-54.
(21) Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005 June 23;352(25):2598-608.
(22) Herold KC, Bluestone JA, Montag AG, Parihar A, Wiegner A, Gress RE, Hirsch R. Prevention of autoimmune diabetes with nonactivating anti-CD3 monoclonal antibody. diab 1992;41(3):385-91.
(23) Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002 May 30;346(22):1692-8.
(24) Ogawa N, List JF, Habener JF, Maki T. Cure of overt diabetes in NOD mice by transient treatment with anti-lymphocyte serum and exendin-4. diab 2004 July;53(7):1700-5.
(25) Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol 2005 September 1;175(5):3053-9.
(26) Solvason N, Lou YP, Peters W, Evans E, Martinez J, Ramirez U, Ocampo A, Yun R, Ahmad S, Liu E, Yu L, Eisenbarth G, Leviten M, Steinman L, Garren H. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J Immunol 2008 December 15;181(12):8298-307.
(27) Kodama S, Kuhtreiber W, Fujimura S, Dale EA, Faustman DL. Islet regeneration during the reversal of autoimmune diabetes in NOD mice. Science 2003 November 14;302(5648):1223-7.
(28) Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. diab 2005 September;54(9):2596-601.
(29) Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. diab 1978;27(Suppl):251-61.
(30) Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab 2008 April 29;.
(31) Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A, Martell R, Rodger NW, Graffenried BV, Wolfe BMJ. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science 1984;223(4643):1362-7.
(32) Chase HP, Butler-Simon N, Garg SK, Hayward A, Klingensmith GJ, Hamman RF, O'Brien D. Cyclosporine A for the treatment of new-onset insulin-dependent diabetes mellitus. peds 1990;85(3):241-5.
(33) Carel J-C, Boitard C, Eisenbarth G, Bach J, Bougnères P. Cyclosporine delays but does not prevent clinical onset in glucose intolerant pre-type I diabetic children. J Autoimmun 1996;9:739-45.
(34) Tuvemo T, Frisk G, Friman G, Ludvigsson J, Diderholm H. IgM against Coxsackie B viruses in children developing Type I diabetes mellitus - a seven year retrospective study. Diabetes Res 1988;9:125-9.
(35) Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet Transplantation in Seven Patients with Type 1 Diabetes Mellitus Using a Glucocorticoid-free Immunosuppressive Regimen. N Engl J Med 2000 July 27;343(4):230-8.
(36) Ryan EA, Lakey JR, Rajotte RV, Korbutt GS, Kin T, Imes S, Rabinovitch A, Elliott JF, Bigam D, Warnock GL, Larsen I, Shapiro AM, Kneteman NM. Clinical outcomes and insulin secretion after islet transplantation with the Edmonton Protocol. diab 2001;50(4):710-9.
(37) Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. diab 2005 July;54(7):2060-9.
(38) Vu MD, Qi S, Xu D, Wu J, Peng J, Daloze P, Sehgal S, Leduc B, Chen H. Synergistic effects of mycophenolate mofetil and sirolimus in prevention of acute heart, pancreas, and kidney allograft rejection and in reversal of ongoing heart allograft rejection in the rat. Transplantation 1998 December 27;66(12):1575-80.
(39) Hao L, Chan S-M, Lafferty KJ. Mycophenolate mofetil can prevent the development of diabetes in BB rats. Ann N Y Acad Sci 1994;696(11):328-32.
(40) Buckingham BA, Sandborg CI. A randomized trial of methotrexate in newly diagnosed patients with type 1 diabetes mellitus. Clin Immunol 2000 August;96(2):86-90.
(41) Hayward AR, Shriber M. Reduced incidence of insulitis in NOD mice following anti-CD3 injection: requirement for neonatal injection. J Autoimmun 1992;5:59-67.
(42) Chatenoud L, Bach JF. Regulatory T cells in the control of autoimmune diabetes: the case of the NOD mouse. Int Rev Immunol 2005 May;24(3-4):247-67.
(43) Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 2007 August;7(8):622-32.
(44) Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003 September;9(9):1202-8.
(45) Begum S, Chen W, Herold KC, Papaioannou VE. Remission of type 1 diabetes following anti-CD3 antibody treatment and transplantation of embryonic pancreatic precursors. Endocrinology 2009 July 9.
(46) Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A Single Course of Anti-CD3 Monoclonal Antibody hOKT3{gamma}1(Ala-Ala) Results in Improvement in C-Peptide Responses and Clinical Parameters for at Least 2 Years after Onset of Type 1 Diabetes. Diabetes 2005 June;54(6):1763-9.
(47) Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P, Sayre P, Bianchine P, Wong E, Seyfert-Margolis V, Bourcier K, Bluestone JA. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol 2009 August;132(2):166-73.
(48) Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005 June 23;352(25):2598-608.
(49) Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A Single Course of Anti-CD3 Monoclonal Antibody hOKT3{gamma}1(Ala-Ala) Results in Improvement in C-Peptide Responses and Clinical Parameters for at Least 2 Years after Onset of Type 1 Diabetes. diab 2005 June;54(6):1763-9.
(50) Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8 T cell population and induces CD8CD25 Tregs. J Clin Invest 2005 October 1;115(10):2904-13.
(51) Kaufman A, Herold KC. Anti-CD3 mAbs for treatment of type 1 diabetes. Diabetes Metab Res Rev 2009 May;25(4):302-6.
(52) Ablamunits V, Bisikirska BC, Herold KC. Human regulatory CD8 T cells. Ann N Y Acad Sci 2008 December;1150:234-8.
(53) Bresson D, Togher L, Rodrigo E, Chen YL, Bluestone JA, Herold KC, von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. Journal of Clinical Investigation 2006;116(5):1371-81.
(54) Shah SC, Malone JI, Simpson NE. A randomized trial of intensive insulin therapy in newly diagnosed insulin-dependent diabetes mellitus. N Engl J Med 1989;320(9):550-4.
(55) Palmer JP, Fleming GA, Greenbaum CJ, Herold KC, Jansa LD, Kolb H, Lachin JM, Polonsky KS, Pozzilli P, Skyler JS, Steffes MW. C-Peptide Is the Appropriate Outcome Measure for Type 1 Diabetes Clinical Trials to Preserve beta-Cell Function: Report of an ADA Workshop, 21-22 October 2001. diab 2004 January;53(1):250-64.
(56) Maruyama T, Tanaka S, Shimada A, Funae O, Kasuga A, Kanatsuka A, Takei I, Yamada S, Harii N, Shimura H, Kobayashi T. Insulin intervention in slowly progressive insulin-dependent (type 1) diabetes mellitus. J Clin Endocrinol Metab 2008 June;93(6):2115-21.
(57) Qin HY, Singh B. BCG vaccination prevents insulin-dependent diabetes mellitus (IDDM) in NOD mice after disease acceleration with cyclophosphamide. J Autoimmun 1997 June;10(3):271-8.
(58) Harada M, Kishimoto Y, Makino S. Prevention of overt diabetes and insulitis in NOD mice by a single BCG vaccination. diab 1990;8:85-90.
(59) Allen HF, Klingensmith GJ, Jensen P, Simoes E, Hayward A, Chase HP. Effect of BCG vaccination on new-onset insulin-dependent diabetes mellitus: A randomized clinical study. Diab care 1998;22(10):1703-7.
(60) Huppmann M, Baumgarten A, Ziegler AG, Bonifacio E. Neonatal Bacille Calmette-Guerin vaccination and type 1 diabetes. Diab care 2005 May;28(5):1204-6.
(61) Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med 2008 December 25;359(26):2849-50.
(62) Warram JH, Krolewski AS, Gottlieb MS, Kahn CR. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N Engl J Med 1984;311(3):149-52.
(63) Ludvigsson J, Lindblom B. Human lymphocyte antigen DR types in relation to early clinical manifestations in diabetic children. Pediatr Res 1984;18(12):1239-41.
(64) Aly TA, Ide A, Jahromi MM, Barker JM, Fernando MS, Babu SR, Yu L, Miao D, Erlich HA, Fain PR, Barriga KJ, Norris JM, Rewers MJ, Eisenbarth GS. Extreme Genetic Risk for Type 1A Diabetes. Proc Natl Acad Sci USA 2006 September 19;103(38):14074-9.
(65) Baschal EE, Aly TA, Babu SR, Fernando MS, Yu L, Miao D, Barriga KJ, Norris JM, Noble JA, Erlich HA, Rewers MJ, Eisenbarth GS. HLA-DPB1*0402 Protects Against Type 1A Diabetic Autoimmunity in the Highest Risk DR3-DQB1*0201/DR4-DQB1*0302 DAISY Population. diab 2007 May 18;56(9):2405-9.
(66) Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007 October 23;104(43):17040-5.
(67) Krischer JP, Cuthbertson DD, Yu L, Orban T, Maclaren N, Jackson R, Winter WE, Schatz DA, Palmer JP, Eisenbarth GS. Screening strategies for the identification of multiple antibody-positive relatives of individuals with type 1 diabetes. J Clin Endocrinol Metab 2003 January;88(1):103-8.
(68) Morel PA, Dorman JS, Todd JA, McDevitt HO, Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects against type I diabetes: a family study. Proc Natl Acad Sci USA 1988;85(21):8111-5.
(69) Baisch JM, Weeks T, Giles R, Hoover M, Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in insulin- dependent diabetes mellitus. N Engl J Med 1990;322(26):1836-41.
(70) Reijonen H, Ilonen J, Knip M, Akerblom HK. HLA-DQB1 alleles and the absence of Asp 57 as susceptibility factors of IDDM in Finland. diab 1991;40:1640-4.
(71) Bingley PJ, Bonifacio E, Gale EAM. Can we really predict IDDM? diab 1993;42:213-20.
(72) Kupila A, Muona P, Simell T, Arvilommi P, Savolainen H, Hamalainen AM, Korhonen S, Kimpimaki T, Sjoroos M, Ilonen J, Knip M, Simell O. Feasibility of genetic and immunological prediction of type I diabetes in a population-based birth cohort. diabetol 2001 March;44(3):290-7.
(73) Field LL. Genetic linkage and association studies of Type I diabetes: challenges and rewards. diabetol 2002 January;45(1):21-35.
(74) Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009 April 16;360(16):1646-54.
(75) Bonifacio E, Bingley PJ, Shattock M, Dean BM, Dunger D, Gale EAM, Bottazzo GF. Quantification of islet-cell antibodies and prediction of insulin-dependent diabetes. Lancet 1990;335:147-9.
(76) Riley WJ, Maclaren NK, Krischer J, Spillar RP, Silverstein JH, Schatz DA, Schwartz S, Malone J, Shah S, Vadheim C, Rotter JL. A prospective study of the development of diabetes in relatives of patients with insulin-dependent diabetes. N Engl J Med 1990;323(17):1167-72.
(77) Karjalainen JK. Islet cell antibodies as predictive markers for IDDM in children with high background incidence of disease. diab 1990;39(9):1144-50.
(78) Yu L, Cuthbertson DD, Maclaren N, Jackson R, Palmer J, Orban T, Eisenbarth GS, Krischer J. Expression of GAD65 and Islet Cell Antibody (ICA512) autoantibodies amongst cytoplasmic ICA+ relatives is associated with eligibility for Diabetes Prevention Trial Type-1. diab 2001;50:1735-40.
(79) Chase HP, Garg SK, Butler-Simon N, Klingensmith G, Norris L, Ruskey CT, O'Brien D. Prediction of the course of pre-type I diabetes. J Pediatr 1991;118(6):838-41.
(80) Bruining GJ, Molenaar JL, Grobbee DE, Hofman A, Scheffer GJ, Bruining HA, DeBruyn AM, Valkenburg HA. Ten-year follow-up study of islet-cell antibodies and childhood diabetes mellitus. Lancet 1989;1(8647):1100-3.
(81) Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002 May 30;346(22):1685-91.
(82) Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996;45(7):926-33.
(83) Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest 2004 August;114(4):589-97.
(84) Bonifacio E, Boitard C, Gleichmann H, Shattock MA, Molenaar JL, Bottazzo G-F. Assessment of precision, concordance, specificity, and sensitivity of islet cell antibody measurements in 41 assays. diabetol 1990;33:731-6.
(85) Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983;222(4630):1337-9.
(86) Mcevoy RC, Witt ME, Ginsberg-Fellner F, Rubinstein P. Anti-insulin antibodies in children with type I diabetes mellitus. Genetic regulation of production and presence at diagnosis before insulin replacement. diab 1986;35(6):634-41.
(87) Baekkeskov S, Aanstoot H-J, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, DeCamilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase [published erratum appears in Nature 1990 Oct 25;347(6295):782]. Nature 1990;347(6289):151-6.
(88) Atkinson MA, Maclaren NK, Scharp DW, Lacy PE, Riley WJ. 64,000 Mr autoantibodies as predictors of insulin-dependent diabetes. Lancet 1990;335(8702):1357-60.
(89) Barmeier H, McCulloch DK, Neifing JL, Warnock G, Rajotte RV, Palmer JP, Lernmark Å. Risk for developing type I (insulin-dependent) diabetes mellitus and presence of islet 64K antibodies. diabetol 1991;34:727-33.
(90) Hawa M, Fava D, Medici F, Deng Y, Notkins A, De Mattia G, Leslie R. Antibodies to IA-2 and GAD65 in type 1 and type 2 diabetes. Diab care 2000;23(2):228.
(91) Srikanta S, Ganda OP, Gleason RE, Jackson RA, Soeldner JS, Eisenbarth GS. Pre-type I diabetes. Linear loss of beta cell response to intravenous glucose. diab 1984;33(8):717-20.
(92) Chase HP, Voss MA, Butler-Simon N, Hoops S, O'Brien D, Dobersen MJ. Diagnosis of pre-type I diabetes. J Pediatr 1987;111(6 Pt 1):807-12.
(93) Keskinen P, Korhonen S, Kupila A, Veijola R, Erkkila S, Savolainen H, Arvilommi P, Simell T, Ilonen J, Knip M, Simell O. First-phase insulin response in young healthy children at genetic and immunological risk for Type I diabetes. diabetol 2002 December;45(12):1639-48.
(94) Ad Hoc Expert Committee. Prevention of type 1 diabetes mellitus. Diab care 1990;13:1026-7.
(95) Bingley PJ, Colman P, Eisenbarth GS, Jackson RA, McCulloch DK, Riley WJ, Gale EAM. Standardization of IVGTT to predict IDDM. Diab care 1992;15:1313-6.
(96) Allen HF, Jeffers BW, Klingensmith GJ, Chase HP. First-phase insulin release in normal children. J Pediatr 1993;123(5):733-8.
(97) Stene LC, Barriga K, Hoffman M, Kean J, Klingensmith G, Norris JM, Erlich HA, Eisenbarth GS, Rewers M. Normal but increasing hemoglobin A1c levels predict progression from islet autoimmunity to overt type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY). Pediatr Diabetes 2006 October;7(5):247-53.
(98) Ad Hoc Expert Committee. Position statement: prevention of type 1 diabetes mellitus. Diabetes 39, 1151-1152. 1990.
Ref Type: Journal (Full)
(99) Barker JM, Goehrig SH, Barriga K, Hoffman M, Slover R, Eisenbarth GS, Norris JM, Klingensmith GJ, Rewers M. Clinical characteristics of children diagnosed with type 1 diabetes through intensive screening and follow-up. Diab care 2004 June;27(6):1399-404.
(100) Sosenko JM, Palmer JP, Greenbaum CJ, Mahon J, Cowie C, Krischer JP, Chase HP, White NH, Buckingham B, Herold KC, Cuthbertson D, Skyler JS. Patterns of metabolic progression to type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 2006 March;29(3):643-9.
(101) Bingley PJ, Mahon JL, Gale EA. Insulin resistance and progression to type 1 diabetes in the European Nicotinamide Diabetes Intervention Trial (ENDIT). Diabetes Care 2008 January;31(1):146-50.
(102) Bingley PJ, Gale EA. Progression to type 1 diabetes in islet cell antibody-positive relatives in the European Nicotinamide Diabetes Intervention Trial: the role of additional immune, genetic and metabolic markers of risk. diabetol 2006 March 3.
(103) Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004 March 20;363(9413):925-31.
(104) Skyler JS, Brown D, Chase HP, Collier E, Cowie C, Eisenbarth GS, Fradkin J, Grave G, Greenbaum C, Jackson RA, Kaufman FR, Krischer JP, Marks JB, Palmer JP, Ricker A, Schatz DA, Wilson D, Winter WE, Wolfsdorf J, Zeidler A, Dickler H, Eastman RC, Maclaren NK, Malone JI, Robertson PR et al. Effects of insulin in relatives of patients with type 1 diabetes mellitus. New Engl J Med 2002;346(22):1685-1691B.
(105) Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diab care 2005 May;28(5):1068-76.
(106) Nanto-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, Korhonen S, Erkkola R, Sipila JI, Haavisto L, Siltala M, Tuominen J, Hakalax J, Hyoty H, Ilonen J, Veijola R, Simell T, Knip M, Simell O. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet 2008 November 15;372(9651):1746-55.
(107) Kolb H, Burkart V. Nicotinamide in type 1 diabetes. Mechanism of action revisited. Diab care 1999 March;22 Suppl 2:B16-B20.
(108) Yamada K, Nonaka K, Miyazaki A, Toyoshima H, Tarui S. Preventive and therapeutic aspects of large dose nicotinamide injections on diabetes associated with insulitis: an observation in ion injection of alloxan and streptozotocin on islet proinsulin synthesis. diab 1982;31:749-53.
(109) Uchigata Y, Yamamoto H, Nagai H, Okamoto H. Effect of poly(ADP-ribose) synthetase inhibitor administration to rats before and after injection of alloxan and streptozotocin on islet proinsulin synthesis. diab 1983;32(4):316-8.
(110) LeDoux SP, Hall CR, Forbes PM, Patton NJ, Wilson GL. Mechanisms of nicotinamide and thymidine protection from alloxan and streptozotocin toxicity. diab 1988;37(8):1015-9.
(111) Kolb H, Burkart V, Appels B, Hanenberg H, Kantwerk-Funke G, Kiesel U, Funda J, Schraermeyer U, Kolb-Bachofen V. Essential contribution of macrophages to islet cell destruction in vivo and in vitro. J Autoimmun 1990;3(Suppl 1):117-20.
(112) Yamada K, Miyajima E, Nonaka K. Inhibition of cytokine-induced MHC class II but not class I molecule expression on mouse islet cells by niacinamide and 3- aminobenzamide. diab 1990;39(9):1125-30.
(113) Pociot F, Reimers JI, Andersen HU. Nicotinamide - biological actions and therapeutic potential in diabetes prevention. diabetol 1993;36:574-6.
(114) Visalli N, Cavallo MG, Signore A, Baroni MG, Buzzetti R, Fioriti E, Mesturino C, Fiori R, Lucentini L, Matteoli MC, Crino A, Corbi S, Spera S, Teodonio C, Paci F, Amoretti R, Pisano L, Suraci C, Multari G, Sulli N, Cervoni M, De Mattia G, Faldetta MR, Boscherini B, Bitti ML et al. A multi-centre randomized trial of two different doses of nicotinamide in patients with recent-onset type 1 diabetes (the IMDIAB VI). Diabetes Metab Res Rev 1999 May;15(3):181-5.
(115) Chase HP, Butler-Simon N, Garg S, McDuffie M, Hoops SL, O'Brien D. A trial of nicotinamide in newly diagnosed patients with type 1 (insulin-dependent) diabetes mellitus. diabetol 1990;33:444-6.
(116) Elliott RB, Chase HP. Prevention or delay of type I (insulin-dependent) diabetes mellitus in children using nicotinamide. diabetol 1991;34(5):362-5.
(117) Gale EA. Theory and practice of nicotinamide trials in pre-type 1 diabetes. J Pediatr Endocrinol Metab 1996 May;9(3):375-9.
(118) World Health Organization. Report of a WHO Study Group. Diabetes Mellitus. Technical Report Series 727, 1-99. 1985.
Ref Type: Generic
(119) Gale EA. Intervening before the onset of Type 1 diabetes: baseline data from the European Nicotinamide Diabetes Intervention Trial (ENDIT). diabetol 2003 March;46(3):339-46.
(120) Gottlieb PA, Handler ES, Appel MC, Greiner DL, Mordes JP, Rossini AA. Insulin treatment prevents diabetes mellitus but not thyroiditis in RT 6-depleted diabetes resistant BB/Wor rats. diabetol 1991;34(5):296-300.
(121) Gottlieb PA, Rossini AA. The BB rat models of IDDM. In: Cohen IR, Miller A, editors. Autoimmune Disease Models: A Guidebook. Academic Press; 1994. p. 163-74.
(122) Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000 April;12(4):431-40.
(123) Roncarolo MG, Levings MK. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr Opin Immunol 2000 December;12(6):676-83.
(124) Weiner HL. Oral tolerance, an active immunologic process mediated by multiple mechanisms. J Clin Invest 2000 October;106(8):935-7.
(125) Bergerot I, Arreaza GA, Cameron MJ, Burdick MD, Strieter RM, Chensue SW, Chakrabarti S, Delovitch TL. Insulin B-chain reactive CD4+ regulatory T-cells induced by oral insulin treatment protect from type 1 diabetes by blocking the cytokine secretion and pancreatic infiltration of diabetogenic effector T-cells. diab 1999 September;48(9):1720-9.
(126) Ploix C, Bergerot I, Durand A, Czerkinsky C, Holmgren J, Thivolet C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. diab 1999 November;48(11):2150-6.
(127) French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, Georgiou HM, Kay TW, Harrison LC, Lew AM. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. diab 1996;46:34-9.
(128) Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J Exp Med 1996;184(6):2167-74.
(129) Keller RJ, Eisenbarth GS, Jackson RA. Insulin prophylaxis in individuals at high risk of type I diabetes. Lancet 1993;341(8850):927-8.
(130) Karounos DG, Bryson JS, Cohen DA. Metabolically inactive insulin analog prevents type I diabetes in prediabetic NOD mice. J Clin Invest 1997 September 15;100(6):1344-8.
(131) Effects of Oral Insulin in Relatives of Patients With Type 1 Diabetes: The Diabetes Prevention Trial-Type 1. Diab care 2005 May;28(5):1068-76.
(132) Bluestone JA, Matthews JB. The immune tolerance network--an NIH/JDF-supported initiative to bring tolerance research into the clinic: a major new resource for clinical immunologists. Clin Immunol 2000 September;96(3):171-3.
(133) Kimpimaki T, Kulmala P, Savola K, Vahasalo P, Reijonen H, Ilonen J, Akerblom HK, Knip M. Disease-associated autoantibodies as surrogate markers of type 1 diabetes in young children at increased genetic risk. Childhood Diabetes in Finland Study Group [In Process Citation]. J Clin Endocrinol Metab 2000 Mar ;85 (3 ):1126 -32 2000;85(3):1126-32.
(134) Norris JM, Beaty B, Klingensmith G, Yu L, Hoffman M, Chase HP, Erlich HA, Hamman RF, Eisenbarth GS, Rewers M. Lack of association between early exposure to cow's milk protein and b-cell autoimmunity: Diabetes Autoimmunity Study in the Young (DAISY). JAMA 1996;276:609-14.
(135) Lempainen J, Vaarala O, Makela M, Veijola R, Simell O, Knip M, Hermann R, Ilonen J. Interplay between PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun 2009 May 25.
(136) Alleva DG, Crowe PD, Jin L, Kwok WW, Ling N, Gottschalk M, Conlon PJ, Gottlieb PA, Putnam AL, Gaur A. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest 2001;107(2):173-80.
(137) Abiru N, Maniatis AK, Yu L, Miao D, Moriyama H, Wegmann D, Eisenbarth GS. Peptide and MHC specific breaking of humoral tolerance to native insulin with the B:9-23 peptide in diabetes prone and normal mice. diab 2001 June;50:1274-81.
(138) Tisch R, Wang B, Serreze DV. Induction of glutamic acid decarboxylase 65-specific Th2 cells and suppression of autoimmune diabetes at late stages of disease is epitope dependent. J Immunol 1999 August 1;163(3):1178-87.
(139) Tisch R, Liblau RS, Yang X-D, Liblau P, McDevitt HO. Induction of GAD65-specific regulatory T-cells inhibits ongoing autoimmune diabetes in nonobese diabetic mice. diab 1998;47:894-9.
(140) Elias D, Cohen IR. Treatment of autoimmune diabetes and insulitis in NOD mice with heat shock protein 60 peptide p277. diab 1995;44(9):1132-8.
(141) Ablamunits V, Elias D, Reshef T, Cohen IR. Islet T cells secreting IFN-gamma in NOD mouse diabetes: arrest by p277 peptide treatment. J Autoimmun 1998 February;11(1):73-81.
(142) Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, Van-Krinks C, Lozanoska-Ochser B, Marquesini L, Brown S, Wong FS, Dayan CM, Peakman M. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol 2009 February;155(2):156-65.
(143) Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol 1994;24(8):1853-7.
(144) Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23). Proc Natl Acad Sci USA 1996;93(2):956-60.
(145) Crawford M, Daniel D, Wegmann D, Yang H, Gill RG. Autoimmune islet damage mediated by insulin-specific T cells. Transplant Proc 1997;29(1-2):758-9.
(146) Liu E, Moriyama H, Abiru N., Miao D, Yu L, Taylor RM, Finkelman F, Eisenbarth GS. Anti-peptide autoantibodies and fatal anaphylaxis in NOD mice in response to insulin self-peptides B:9-23 and B:13-23. J Clin Invest 2002;110(7):1021-7.
(147) Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci U S A 2002 April 16;99(8):5539-44.
(148) Weber SE, Harbertson J, Godebu E, Mros GA, Padrick RC, Carson BD, Ziegler SF, Bradley LM. Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. Journal of Immunology 2006;176(8):4730-9.
(149) Hummel S, Hummel M, Banholzer J, Hanak D, Mollenhauer U, Bonifacio E, Ziegler AG. Development of autoimmunity to transglutaminase C in children of patients with type 1 diabetes: relationship to islet autoantibodies and infant feeding. diabetol 2007 February;50(2):390-4.
(150) Ludvigsson J, Faresjo M, Hjorth M, Axelsson S, Cheramy M, Pihl M, Vaarala O, Forsander G, Ivarsson S, Johansson C, Lindh A, Nilsson NO, Aman J, Ortqvist E, Zerhouni P, Casas R. GAD Treatment and Insulin Secretion in Recent-Onset Type 1 Diabetes. N Engl J Med 2008 October 8;359:1909-20.
(151) Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR. Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet 2001 November 24;358(9295):1749-53.
(152) Schloot NC, Meierhoff G, Lengyel C, Vandorfi G, Takacs J, Panczel P, Barkai L, Madacsy L, Oroszlan T, Kovacs P, Suto G, Battelino T, Hosszufalusi N, Jermendy G. Effect of heat shock protein peptide DiaPep277 on beta-cell function in paediatric and adult patients with recent-onset diabetes mellitus type 1: two prospective, randomized, double-blind phase II trials. Diabetes Metab Res Rev 2007 May;23(4):276-85.
(153) Eldor R, Kassem S, Raz I. Immune modulation in type 1 diabetes mellitus using DiaPep277: a short review and update of recent clinical trial results. Diabetes Metab Res Rev 2009 May;25(4):316-20.
(154) Bowman M, Atkinson MA. Heat shock protein therapy fails to prevent diabetes in NOD mice. diabetol 2002 September;45(9):1350-1.
(155) Flohe SB, Bruggemann J, Lendemans S, Nikulina M, Meierhoff G, Flohe S, Kolb H. Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J Immunol 2003 March 1;170(5):2340-8.
(156) Habich C, Kempe K, van der ZR, Burkart V, Kolb H. Different heat shock protein 60 species share pro-inflammatory activity but not binding sites on macrophages. FEBS Lett 2003 January 2;533(1-3):105-9.
(157) Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 2000 January 15;164(2):558-61.
(158) Cohen IR. Peptide therapy for Type I diabetes: the immunological homunculus and the rationale for vaccination. diabetol 2002 October;45(10):1468-74.
(159) Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med 2002 December 16;196(12):1645-51.
(160) Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 2008 March;9(3):239-44.
(161) Earle KE, Tang Q, Zhou X, Liu W, Zhu S, Bonyhadi ML, Bluestone JA. In vitro expanded human CD4+CD25+ regulatory T cells suppress effector T cell proliferation. Clinical Immunology 2005;115(1):3-9.
(162) Game DS, Cao X, Jiang S. Regulatory T cells: the cunning fox and its clinical application. Sci Signal 2008;1(51):mr3.
(163) Trucco M, Giannoukakis N. Immunoregulatory dendritic cells to prevent and reverse new-onset Type 1 diabetes mellitus. Expert Opin Biol Ther 2007 July;7(7):951-63.
(164) Giannoukakis N, Phillips B, Trucco M. Toward a cure for type 1 diabetes mellitus: diabetes-suppressive dendritic cells and beyond. Pediatr Diabetes 2008 June;9(3 Pt 2):4-13.
(165) Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simoes BP, Martinez EZ, Foss MC, Burt RK, Voltarelli JC. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2009 April 15;301(15):1573-9.
(166) Eisenbarth GS, Srikanta S, Jackson R, Rabinowe S, Dolinar R, Aoki T, Morris MA. Anti-thymocyte globulin and prednisone immunotherapy of recent onset type I diabetes mellitus. Diabetes Res 1985;2(6):271-6.
(167) Wucherpfennig KW, Eisenbarth GS. Type 1 Diabetes. Nature Immunology 2001;2(7):1-3.
(168) Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, Santamaria P, Tan R. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 2003 January;111(2):217-23.
(169) Nepom GT. Approaching the Heisenberg principle of immunology. Clin Immunol 2008 July 18.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12531877&query_hl=161