Type 1 Diabetes: Cellular, Molecular & Clinical Immunology
Chapter 2 - The Pancreatic Beta-Cell
Suparna A. Sarkar, 3/1/2012 Update of chapter by Kirstine Juhl, John C. Hutton
and George S. Eisenbarth
Human Fetal Pancreas Development slide set Updated 03/12
Introduction
The two most common forms of diabetes in man (Type 1A and Type 2) have very
different etiologies and different clinical presentation1. Nevertheless, the underlying loss of islet beta
cell function has similar consequences in terms of glycemic control and the
emergence of long-term complications. type 1 Diabetes (T1D) is a polygenic
T-cell dependent autoimmune disease, characterized by the selective destruction
of the ß-cells of the islets of Langerhans1-6 and that susceptible individuals have inherent defects in critical immunomodulatory
mechanisms7 that increase the risk of a pathogenic rather than
protective immune response to self6, 8, 9. Type 2 Diabetes is typically linked to dysmetabolism or metabolic
syndrome and the presence of insulin resistance,
however a large subset of T1D patients routinely exhibits insulin resistance10-13 contributing to the metabolic distress in islets. With the rising incidence of T1D and
T2D, it is now being argued that both T1D and T2D are essentially disorders of
altered insulin resistance set against the backdrop of genetic susceptibility14 and the inflammatory process; in T1D brought about
by the autoimmune component of the disease process15.
In type 1A
(autoimmune diabetes) the loss of beta cells is often close to absolute with
less than 1% of beta cells remaining in patients with long-term diabetes16-18 with prolonged C peptide production19. In most
patients with almost no remaining beta cells, essentially all of the islets are
devoid of beta cells while islets contain cells expressing glucagon and
somatostatin. Such islets are termed pseudoatrophic islets20. Nevertheless, some beta cells remain often as scattered single cells
in the parenchyma and ducts. In a
small subset of patients, even with long-term type 1A diabetes, significant
C-peptide is present and lobules of pancreas remain where all the islets
contain beta cells and appear essentially normal in terms of expression of
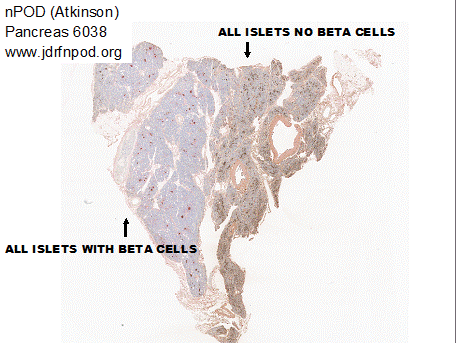
insulin while the rest of the pancreas is devoid of beta cells in islets20-22. The figure below illustrates a section of one such pancreas from the
nPOD collection (jdrfnpod.org) where the pancreatic lobule on the right is
stained dark and at higher power one can observe that all of the islets in this
lobule lack insulin23. In
contrast to the lobule on the left, all of the islets contain insulin. The dark staining of the right lobule
likely results from pancreatic acinar atrophy that occurs with severe loss of
pancreatic insulin. Shrinkage in
overall pancreatic mass in patients with type 1 diabetes has long been noted24-26. Analysis of decreased pancreatic volume was recently combined with
imaging of iron particle pancreatic accumulation to help distinguish patients
with type 1 diabetes from normal controls27-29.
 In fact, C-peptide secretion
in long-standing diabetic patients has now been explained by two different
patterns of beta cell survival, which possibly reflect different subsets of
type 1 diabetes. In a recent study20 associated Pattern A with type 1A diabetes that
histologically had lobular retention of islet areas with “abnormal’ beta cells
producing the apoptosis inhibitor BIRC5 (survivin) and HLA class I. In pattern
B, 100% of all islets contained normal-appearing but quantitatively reduced
beta cells without survivin or HLA class I. Baculoviral IAP
repeat 5 BiRC530 is an apoptosis inhibitor that is produced in the
beta cells of fetal human pancreas31, but not in adult islets. It is also found in the beta cells in areas
of pancreatitis32. The presence of survivin,
in all surviving islet beta cells Pattern A patients, may result from 1)
inflammatory changes that did not result in beta cell destruction of a subset
of islets or 2) be protected from destruction and further lymphocytic
infiltration. An alternative hypothesis extended by the authors is that lobular
regions with beta cells of Pattern A pancreas represent areas of beta cell
regeneration. Although the sudden onset
of type 1A belies the fact that the underlying loss of beta cell mass is the
culmination of many years of gradual and progressive loss of beta cells in the
face of autoimmune attack which is first evident with the appearance of
autoantibodies to islet proteins in the preceding years (see other chapters)33-38. In the NOD mouse the infiltration of the islets with immune and
inflammatory cells that initiates the disease first appears in the islets of
the pancreatic periphery, affects a subpopulation of islets and is possibly
benign or at least kept in check by the presence of regulatory T cells39-42. The invasive insulitis seen in NOD mice closer to disease onset may
reflect a change in the balance of destructive and protective responses in
favor of the former. The histological changes in man are comparatively mild and
may reflect the slower progression of the disease or possibly a different
immune process. The islet tends to be viewed as the source of autoantigen that
supports or initiates the immune attack and ultimately the victim of the crime.
In fact, C-peptide secretion
in long-standing diabetic patients has now been explained by two different
patterns of beta cell survival, which possibly reflect different subsets of
type 1 diabetes. In a recent study20 associated Pattern A with type 1A diabetes that
histologically had lobular retention of islet areas with “abnormal’ beta cells
producing the apoptosis inhibitor BIRC5 (survivin) and HLA class I. In pattern
B, 100% of all islets contained normal-appearing but quantitatively reduced
beta cells without survivin or HLA class I. Baculoviral IAP
repeat 5 BiRC530 is an apoptosis inhibitor that is produced in the
beta cells of fetal human pancreas31, but not in adult islets. It is also found in the beta cells in areas
of pancreatitis32. The presence of survivin,
in all surviving islet beta cells Pattern A patients, may result from 1)
inflammatory changes that did not result in beta cell destruction of a subset
of islets or 2) be protected from destruction and further lymphocytic
infiltration. An alternative hypothesis extended by the authors is that lobular
regions with beta cells of Pattern A pancreas represent areas of beta cell
regeneration. Although the sudden onset
of type 1A belies the fact that the underlying loss of beta cell mass is the
culmination of many years of gradual and progressive loss of beta cells in the
face of autoimmune attack which is first evident with the appearance of
autoantibodies to islet proteins in the preceding years (see other chapters)33-38. In the NOD mouse the infiltration of the islets with immune and
inflammatory cells that initiates the disease first appears in the islets of
the pancreatic periphery, affects a subpopulation of islets and is possibly
benign or at least kept in check by the presence of regulatory T cells39-42. The invasive insulitis seen in NOD mice closer to disease onset may
reflect a change in the balance of destructive and protective responses in
favor of the former. The histological changes in man are comparatively mild and
may reflect the slower progression of the disease or possibly a different
immune process. The islet tends to be viewed as the source of autoantigen that
supports or initiates the immune attack and ultimately the victim of the crime.
Histopathological
examination of pancreata from diabetic organ donors procured from nPOD was
examined with the goal to provide a foundation for the informed selection of
potential therapeutic targets within the chemokine/receptor family43. CCL5, CCL8, CCL22, CXCL9, CXCL10 and CX3CL1
were the major chemokines transcribed and translated by human islet cells in
response to in vitro inflammatory stimuli. CXCL10 was identified as the
dominant chemokine expressed in vivo in the islet environment of
prediabetic animals and T1D patients, while CCL5, CCL8, CXCL9 and CX3CL1
proteins were present at lower levels in the islets of both species.
Importantly, additional expression of the same chemokines in human acinar
tissues emphasized an underappreciated involvement of the exocrine pancreas in
the natural course of T1D that will require consideration for further T1D
pathogenesis and immune intervention studies.
Undoubtedly, much
more needs to be learned about the reaction of the islet to cytokine mediators
of the immune response and about how the beta cell manages to survive so long
or replenish its population from progenitor cells in the pancreas. Since the
mechanism of autoimmune destruction by effector cells may be mediated by CD4+
cells, and thus indirect, there is also the question of whether the beta cell
is uniquely susceptible to oxygen and nitrogen free radicals or cytokine
mediators of cell death which may account for the fact that other islet cells
exposed to same molecules survive while the beta cell dies.
The focus of the
following review is to discuss the wealth of information regarding the
physiological and pathophysiological responses of the islet to nutrient
secretagogues and pharmacological agents and to emphasize how the beta cell
differs from its neighbors and from other endocrine tissues and how it may
participate in its own demise in type 1 diabetes. The review also illustrates
the challenges faced by investigators wishing to genetically engineer non-b cells for cellular therapy of type 1 diabetes or
wanting to introduce specific genes into the beta cell population to afford it
greater protection from autoimmune attack.
Development of the Human Pancreas
Similar to the mouse pancreas, the human pancreas develops from two
endodermal diverticula, the dorsal and ventral44, which fuses around 56 days post coitum of development45
. The pancreas comprises of 3 important
cell lineages: Endocrine, acinar and ductal (which together make up the exocrine
pancreas). The morphogenesis of the endocrine tissue, however, is unlikely to
be equivalent given the differences in gestation (260 vs 20 days) and the
larger relative volume of the human pancreas46. Human fetal pancreases obtained at gestational ages
9–23 weeks were processed in parallel for immunohistochemistry and gene
expression profiling by Affymetrix microarray47. At 9–11 weeks, the pancreas was made up principally of
mesenchymal tissue interspersed with PDX1 positive branched epithelial
structures containing scattered hormone-negative neurogenin3-positive endocrine
cells. Protoacinar structures marked by carboxy esterase lipase (CEL)
expression were noted by 15–19 weeks, along with clusters of endocrine
cells producing either glucagon or insulin. By 20–23 weeks, vascularized
islet-like structures appeared. Analysis of Ki67 immunoreactivity showed that
the replicative rate of endocrine cells was low and suggested that the
endocrine expansion was derived from hormone-negative precursors. Insulin,
glucagon, somatostatin, ghrelin and pancreatic polypeptide transcripts were
present at 9–10 weeks as confirmed by quantitative PCR and increased
progressively, commensurate with the expansion of endocrine cell volume. The
human equivalent of a mouse endocrine secondary transition was not evident,
neither in terms of morphology nor in dramatic changes in endocrine-specific
transcriptional regulators. By contrast, exocrine genes showed a marked
transition at around 11 weeks, associated with a greater than six-fold
increase in exocrine gene transcripts.
The terminal differentiation of human endocrine tissue into late gestation
and the presence of NEUROG3 are in contrast with findings in the mouse, where
neurog3 is transiently expressed from e12.5–e15.5. This indicates that the
human fetal pancreas could provide an abundant islet precursor cell population
that could be expanded ex vivo for therapeutic transplantation for the
treatment of brittle and unstable type 1 diabetes. The ductal cells also
develop from the PDX1 expressing primordial pancreatic epithelium and expresses
Cytokeratin 19 (CK19), cystic fibrosis transmembrane receptor, DBA lectin,
Carbonic anhydrase 248. (For images of the human fetal pancreas development, please refer to power-point
slides).
Physiology of
the islet of Langerhans
 The endocrine
pancreas is arranged in clusters of secretory cells the Islets of Langerhans
scattered throughout the exocrine glandular tissue (Fig. 1)49, 50. In man, the pancreas contains around one million islets that comprise
1-2% of the total mass of the gland. The islets are separated from the exocrine
tissue by a capsule made up of connective tissue fibers and by glial like cells
and human islets vary in size from less than 50 up to several thousand cells.
Four different endocrine cell types are contained in the islets; beta cells,
which produce insulin and constitute 60-80% of the endocrine cell mass,
glucagon secreting a-cells (10-20%), somatostatin producing d-cells (~5%) and
pancreatic polypeptide secreting PP-cells (<1%)44. The cells within islets are critically
dependent upon a series of transcription factors during embryonic development
and of note, in knockouts of the homeodomain protein nkx2.2, almost all islet
cells are replaced with ghrelin producing cells51. In parallel, in the human
pancreas, the endocrine
genes insulin, glucagon and somatostatin steadily increase from 9–15 weeks,
tapering off by 18–23 weeks47. This was in tandem with other dense core
secretory granule markers such as PCSK1, PCSK1N, PTPRN, SLC30A8, IAPP, CHGA and
CHGB that peak in the later phase of development. Components of the stimulus
secretion coupling machinery such as GCK and GLUT2 (SLC2A4) are however more
variable. The high signal intensity observed for insulin, glucagon and
somatostatin correspond to the abundance of immunopositive cells detected by
immunohistochemistry. Low signals for ghrelin (GHRL) and pancreatic polypeptide
correspond to the lower frequency of these cells in the histological analysis.
The endocrine
pancreas is arranged in clusters of secretory cells the Islets of Langerhans
scattered throughout the exocrine glandular tissue (Fig. 1)49, 50. In man, the pancreas contains around one million islets that comprise
1-2% of the total mass of the gland. The islets are separated from the exocrine
tissue by a capsule made up of connective tissue fibers and by glial like cells
and human islets vary in size from less than 50 up to several thousand cells.
Four different endocrine cell types are contained in the islets; beta cells,
which produce insulin and constitute 60-80% of the endocrine cell mass,
glucagon secreting a-cells (10-20%), somatostatin producing d-cells (~5%) and
pancreatic polypeptide secreting PP-cells (<1%)44. The cells within islets are critically
dependent upon a series of transcription factors during embryonic development
and of note, in knockouts of the homeodomain protein nkx2.2, almost all islet
cells are replaced with ghrelin producing cells51. In parallel, in the human
pancreas, the endocrine
genes insulin, glucagon and somatostatin steadily increase from 9–15 weeks,
tapering off by 18–23 weeks47. This was in tandem with other dense core
secretory granule markers such as PCSK1, PCSK1N, PTPRN, SLC30A8, IAPP, CHGA and
CHGB that peak in the later phase of development. Components of the stimulus
secretion coupling machinery such as GCK and GLUT2 (SLC2A4) are however more
variable. The high signal intensity observed for insulin, glucagon and
somatostatin correspond to the abundance of immunopositive cells detected by
immunohistochemistry. Low signals for ghrelin (GHRL) and pancreatic polypeptide
correspond to the lower frequency of these cells in the histological analysis.
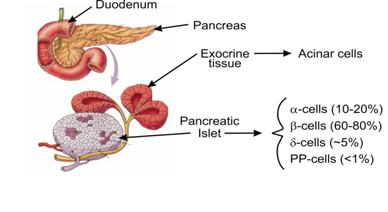
Figure 1. The pancreas is located close to the duodenum. The enlarged
part of the pancreas shows the exocrine acinar cells next to the islet
endocrine cells. The islets consist of four different cell types, which secrete
different peptide hormones. The majority of the islets are beta cells that
secrete insulin. The a-cells secrete glucagon, d-cells secrete somatostatin and
PP cells secrete pancreatic polypeptide.
Most mammalian islets
have a beta cell rich core surrounded by a mantle of alpha-, delta- and
PP-cells with some controversy as to applicability to human islets. Afferent
arterioles enter the beta cell rich medulla where the arterioles split into a
branching system of capillaries that traverse the beta cell mass before
reaching the mantle of a- and d-cells. Thus, the beta cells are the first
endocrine islet cell type to sense blood-borne metabolic changes. Insulin is
known to inhibit glucagon secretion and glucagon to stimulate insulin
secretion. Somatostatin will inhibit the secretion of both insulin and
glucagons. Somatostatin's inhibition of insulin secretion is reported to result
from its inhibition of glucose metabolism of the beta cell (14) and glucagon;
hence the anatomical relationship of the islet cells is critical to the
function of the organ. The islets are innervated by autonomic nerve
fibers, containing both parasympathetic and sympathetic nerves that enter the
islets and terminate in proximity to the endocrine cells.
The architecture of
the human islets is however more heterogeneous. A progressive increase in the endocrine cell
population is evident from 11–23 weeks, reflecting both the progressive
expansion of the pancreatic epithelium and an increase in the density of
endocrine cells within the epithelium47. With increasing fetal age
evidence of formation of endocrine cell clusters especially from 15 weeks
onwards is noted, although solitary endocrine cells were still observed at all
time-points. The endocrine cell clusters at early time-points displayed a
significant homotypic association of either insulin- or glucagon-positive
cells. At later developmental time-points, heterotypic endocrine cell clustering
and the appearance of typical islet-like structures is a feature.
The Beta
Cell as a Neuron-like Cell
Though islet beta
cells are derived from endoderm and not ectoderm they share many properties
with neurons that probably relate to their function in terms of secreting a
peptide hormone stored in dense core secretory granules and having
microvesicles with "neurotransmitters". The neuron-like
properties include presence of microvesicles containing molecules such as GABA
gamma-Aminobutyric acid (GABA)52, an inhibitory neurotransmitter, expression of enzymes such as Glutamic
acid decarboxylase (GAD, both GAD65 and GAD67), IA-2 (ICA512)53, 54, IA-2beta (phogrin)55. Surface expression of complex neuronal/Oligodendrocyte gangliosides
such as GQ gangliosides (e.g. target of monoclonal A2B5) and the targets of
monoclonals 3G556 and R2D657, and expression of type 2 monoamine vesicular
transporters(VMAT2)58, 59 is also seen. These shared
neuronal/neuroendocrine properties are of importance in considering the
pathophysiology of autoimmune beta cell destruction (e.g. though GAD65
autoantibodies are characteristic of type 1 diabetes of man, only beta cells
and not the many other cell types that express GAD65 are destroyed in type 1A
diabetes and in contrast in Stiff Man Syndrome neurologic disease predominates
in association with very high levels of GAD65 autoantibodies) and recently for
the in vivo detection of islets, potentially for determining beta cell
mass. PAM peptidylglycine
alpha-amidating monooxygenase gene encodes a multifunctional protein with two
enzymatically active domains with catalytic activities; peptidylglycine
alpha-hydroxylating monooxygenase (PHM) and peptidyl-alpha-hydroxyglycine
alpha-amidating lyase (PAL)60-62. These catalytic domains work sequentially to catalyze neuroendocrine
peptides to active alpha-amidated products is also found in hypothalamic
magnocellular neurons, the hippocampal formation, and olfactory cortex63 and human endocrine pancreas64. PAM is localized primarily in the secretory granules of alpha cells
not in the beta, PP and Delta cells. Alpha cells secrete GLP1, which possess a
carboxy terminal amide group65 while beta and delta cells that secrete insulin and
somatostatin respectively which are not terminally amidated. Investigators have
utilized [(11)C]Dihydrotetrabenazine ([(11)C]DTBZ) that binds specifically to
VMAT2, with PET scanning to estimate beta cell mass in the BB type 1 diabetes
rat59. It is possible that many of the technologies
developed to interrogate the brain (e.g. MRI spectroscopy) will be applicable
to studies of islet beta cells, if the goal is determination of approximate
mass rather than direct visualization of the relatively small islets, given the
shared biochemistry of neurons and islets.
The
Biosynthesis of Insulin
The islet beta cell is the principal cell in the adult mammal able to
transcribe the insulin gene. The insulin receptor by comparison is widely
distributed even on cells that are not thought to be insulin responsive and in
the case not even exposed to significant concentrations of the hormone. This
may reflect the evolution of the molecule from primarily a neurotransmitter to
an endocrine function66. Of note
a series of rare mutations which result in misfolded insulin and neonatal
diabetes have now been identified67. Beta cell death may be the
result of ER stress68-70.
Insulin mRNA is translated to a pre-pro-insulin peptide, which is rapidly
processed in the rough endoplasmic reticulum to pro-insulin by removal of the
N-term signal peptide. Proinsulin contains the A and B chains of insulin linked
by the C peptide, a structure that helps to align the disulfide bridges that
are generated between the A and B chain. Proinsulin is transported through the
Golgi apparatus and thereafter further processed in the maturing granule to
insulin by excision of the C peptide by the endopeptidases, PC1 and PC2
(prohormone convertases), and carboxypeptidase H. The bioactive insulin
molecule consists of an A and B chain (21 and 30 amino acids, respectively), linked
intramolecularly by disulfide bridges71-77.
Role of Zinc in insulin biosynthesis (Leah Sheridan, PhD)
 Insulin
secretion is tightly regulated by extracellular secretagogues/inhibitors, as
well as intracellular signaling pathways.
It is packaged in dense core vesicles (DCVs) as a hexamer bound to two
Zn2+ ions and remains stable in this configuration at pH5.5 until
fusion of the DCV and plasma membranes during exocytosis78, 79 . The complex is then released into the pH neutral circulation, and
dissociates, allowing both insulin and Zn2+ to act independently.
Insulin
secretion is tightly regulated by extracellular secretagogues/inhibitors, as
well as intracellular signaling pathways.
It is packaged in dense core vesicles (DCVs) as a hexamer bound to two
Zn2+ ions and remains stable in this configuration at pH5.5 until
fusion of the DCV and plasma membranes during exocytosis78, 79 . The complex is then released into the pH neutral circulation, and
dissociates, allowing both insulin and Zn2+ to act independently.
The
Zn2+ content of pancreatic b-cells is one of the highest in the body80. This is evidently due to
its additional role in insulin crystal formation, beyond its various
fundamental roles in protein stability and function, DNA replication, metabolic
enzyme activity, and cellular protection against apoptosis and oxidative stress81, 82. In addition, co-secreted
Zn2+ is believed to play both an autocrine and paracrine role by
activating KATP channels, thereby inhibiting the secretory process83, 84, and regulating alpha cell glucagon secretion84, 85. Zn2+ is also implicated in b-cell death via a paracrine mechanism86. Although it is apparent that
mammalian cells require Zn2+ for many cellular processes, it can
also be toxic. The regulation of intracellular Zn2+ is therefore of
great importance to maintain homeostatic cellular physiology. One such
regulatory mechanism involves controlling the influx and efflux of Zn2+
across membranes, both cellular and subcellular, by Zn2+
transporters.
Mammalian
Zn2+ transporters were first discovered in the mid-1990s, and were
shown to transport Zn2+ and other metal ions into the cytoplasm
either from the extracellular space, or from the organellar lumen. These were
members of the SLC39 (also known as ZIP) family of Zn2+
transporters. Later, the SLC30 (also known as ZnT) family of Zn2+
transporters were identified that worked counter to ZIPs, in that they transport
Zn2+ out of the cytoplasm, either out of the cell into the
extracellular space, or into the lumen of organelles87, 88. Mammals carry genes for 10 homologous Zn2+ export proteins
(ZnT1 through 10). The overall homology of ZnT9, however, to other members of
the ZnT family is very low89, and may not function in Zn2+ transport, but potentially in
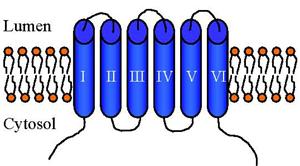
DNA excision-repair90, 91. ZnT8 and ZnT10 were the
latest members to be identified, and akin to ZnT1-7, share the same predicted
structure of six transmembrane (TM)-spanning domains with cytoplasmic amino (N)
and carboxy (C) terminals (see above cartoon), and contain a histidine-rich
intracellular loop between TM IV and V which may act as the Zn2+
binding region. ZnT6 is an exception, in that it retains the prokaryotic
serine-rich loop. Despite the similar topological structure of the ZnT family,
the overall sequence similarities are relatively low, with the highest being
53.5% between ZnT2 and ZnT889. This sequence divergence may reflect the different localizations of
each transporter (i.e. ZnT1-plasma membrane, ZnT2-endosome/lysosome, ZnT3-synaptic
vesicles, etc)92, 93 and correspondingly the different tasks and means of regulation in
which each transporter partakes; a combined bioinformatics/molecular
engineering strategy to identify potential molecular targets of circulating
type 1 diabetic autoantibodies94. In such, they identified
51 candidate islet autoantigens from microarray mRNA expression profiling
experiments on isolated mouse and human pancreatic islets, FACS sorted b-cells and mouse pancreatic tumor cell lines. Among
the candidates, this study identified the Zn2+ transporter ZnT8 as a
novel type 1 diabetes (T1D) autoantigen, and furthermore, localized the epitope
of immunoreactivity to the C-terminus of the protein. Among the other major
known targets of diabetes autoantibodies (insulin, GAD, IA2, and phogrin)
identified by the study, ZnT8 was found to be b-cell specific, displayed moderate to abundant islet
expression, and is associated with the regulated secretory pathway.
The
identification of ZnT8 with roots in the pathogenesis of T1D creates
opportunities for the development of improved diagnostic capabilities as well
as provides an additional strategy for the creation of therapeutic
interventions. Like the other four main autoantigens associated with T1D
studied to date (insulin, GAD, IA2, and phogrin), ZnT8 has been shown to be
associated with the regulated pathway of secretion, having been localized to
intracellular vesicles in HeLa cells, and co-localized with insulin granules in
INS-1 cells and insulin in human islets95, 96. The importance of such an association with the secretion pathway
remains to be elucidated; however, one can speculate that trafficking of these
antigens to and from the surface membrane may play a role in the diabetic
pathology. Due to the specific function of insulin secretion by pancreatic b-cells, proteins specifically related to that
function are potentially more likely to be targeted by the immune system.
Several possible mechanisms come to mind:
1) Proteins associated with DCVs (i.e. insulin, phogrin) that get
trafficked to and from the surface in response to insulin secretagogues, expose
extracellular epitopes that are recognized by T Cell receptors or circulating
antibodies secreted by B cells; 2) Epitopes may be exposed to the immune system
during normal tissue turnover, especially for intracellular proteins (i.e. GAD
and IA2) that would not expose surface epitopes; 3) Recycling of secretory
pathway associated proteins enables access to post-golgi compartments where the
protein could be cut up by proteosomes and presented on the cell surface by MHC
I for recognition by T Cells. These
are only a few of many potential cellular mechanisms by which a protein may
become antigenic. In light of the
recent discovery of ZnT8 and its identification as a T1D autoantigen, it is
essential to investigate its role in pancreatic b-cells to gain insight into its mechanism of antigen
presentation, and furthermore, to generally learn why many other autoantigens
are associated with the DCV (i.e. insulin, CPE and amylin). One study has been
conducted on the functionality of ZnT8 to date, which showed that its
overexpression does not play a role in Zn2+ toxicity, and protected
INS-1 cells, an insulin-secreting cell line, from death during Zn2+
depletion (86). ZnT8 therefore appears to
be a functional channel that actively participates in intracellular Zn2+ transport.
However, much remains to be discovered about this transporter. It has been
shown to co-localize with insulin95, but whether ZnT8 is trafficked to the surface
membrane with insulin upon stimulation, and therefore truly associated with
DCVs remains to be shown. In
addition, if ZnT8 is trafficked to the surface membrane, how long it lingers
there before being re-endocytosed, and where it is trafficked to following its
retrieval is unknown. The processes of DCV exocytosis and endocytosis are well
studied. DCVs undergo regulated exocytosis in
response to signals that elevate cytosolic Ca2+, a process that
must ultimately be coupled to endocytosis of the granule membrane to avoid
significant increases in cell surface area. The concept that exocytosis can occur by
two related, but mechanistically distinct mechanisms (“kiss-and-run” vs.
“complete-collapse”)97-99; has its endocytic counterpart that “kiss-and-run” events are
associated with rapid clathrin-independent retrieval, and “complete-collapse”
with a slower clathrin-dependent mechanism97, 98, 100. In neuroendocrine tissues the type of event that occurs is dictated by
the strength and duration of the stimulus101, and possibly related to the prevailing cytosolic Ca2+ level97, 102. Thus clathrin-independent endocytosis predominates in the initial
phases of the secretory response, while chronic stimulation (> 5 min) leads
to a shift to a mainly clathrin-dependent mechanism and full fusion of DCVs101. Insulin secretion in vivo is
biphasic, which may reflect the release of docked vs. recruited DCVs, changes
in the stimulus intensity as reflected by plasma membrane potential and
cytosolic Ca2+, or indeed the mechanism of granule fusion and
retrieval as discussed above103-105.
Most mammalian
insulin binds Zn co-ordinately forming hexamers and can form large crystalline
arrays which promote condensation and aggregation of the hormone in the acidic
core of the secretory granule. Proinsulin although forming Zn hexamers is
prevented from further aggregation by the highly charged C-peptide, a physical
property that may be important in retaining its solubility in the proximal
elements of the secretory pathway. In the mature secretory granule C peptide
exists in equimolar amounts with insulin in the granule and is co-secreted thus
making it a useful marker for beta cell function in diabetics receiving insulin
therapy. Transcription of the insulin gene is modulated by nutrient
secretagogues, a variety of cytokines and by insulin itself. Insulin signal
transduction enhances transcription of the insulin gene through phosphorylation
of insulin receptor substrate (IRS)-1 and -2, and activation of
phosphatidylinositol-3-kinase (PI3K) and it has been postulated that insulin
plays a feed forward role in insulin biosynthesis ensuring that the stores of
insulin are always replenished106. Exogenous insulin is also shown to hyperpolarize beta cell plasma
membrane in mice by insulin-induced activation of the KATP channels possible
through the PI3K thereby turning off the islet insulin secretion107. Glucose is also shown to stimulate the production of proinsulin
through rapid activation of translation of a pre-formed pool of mRNA108. Given the high rates of proinsulin synthesis and
the need for proper folding of proinsulin within the endoplasmic reticulum it
has been hypothesized that pancreatic beta cells are particularly susceptible
to apoptosis induced by endoplasmic reticulum (ER) stress109. Wolfram syndrome (DIDMOAD: Diabetes Insipidus/Diabetes Mellitus/Optic
atrophy/Nerve Deafness) results from mutations of the WFS1 gene which is a transmembrane
protein of the ER regulated by the unfolded protein response and with a role in
ER calcium transport109.
The
proinflammatory cytokines IL-1b, TNFa and IFNg modulate transcription of the
insulin gene. Culturing human beta cells 48 or 72 h with single cytokines does
not affect the cellular content in insulin or proinsulin whereas the
combination of several cytokines (IL-1β + IFNγ) disproportionally elevates
medium proinsulin levels110. This is caused by a conserved proinsulin synthesis and a slower
hormone conversion possibly contributed to lower expression of PC1 and PC2
convertases. These cytokines mediates signal transduction involving binding to
specific receptors, through MAPK and SAPK and mobilization of diverse
transcription factors: NFkB, AP-1 and STAT-1 involved in beta cell apoptosis 111, 112
Physiological
regulation of islet hormone secretion
The blood glucose level is maintained within a narrow range around 5 to 7 mM in
the fasting state mainly by the combined and reciprocal action of insulin and
glucagon113, 114. Essentially, when the blood glucose concentration is elevated after a
meal, the beta cells are stimulated to secrete insulin. Conversely, when blood
glucose levels are low the a-cells are stimulated to secrete glucagon, which
leads to glycogenolysis and gluconeogenesis in the liver and therefore release
of glucose to the blood. Between meals, when the plasma glucose decreases, the
release of the neurotransmitter norepinephrine and the neuropeptide galanin
from the sympathetic nerves will directly activate glucagon release and inhibit
insulin release115. During a meal or the postprandial state, the parasympathetic nerves
potentiate glucose-induced insulin release from islet beta cells. This is
achieved by the release of the neurotransmitter acetylcholine and the
neuropeptides pituitary adenylate cyclase activating polypeptide (PACAP),
vasoactive intestinal polypeptide (VIP), GLP-1 116-119 and GIP113. The innervation of the islet and its microanatomy is probably
important in the orchestration of these responses. Somatostatin inhibits both
insulin and glucagon secretion and potentially serves as a modulator of islet
hormone release120. However, it is unclear whether it has a physiological role given the
vascular anatomy of the islet in which the secreted products from the a- and
d-cell enter the blood circulation without reaching the beta cells. The
physiological role of islet PP is unknown, but appears not to affect the
secretion of the other islet hormones except a possible inhibition of islet
insulin release121-123. Rare cells within islets secrete ghrelin, with ghrelin primarily
produced in the gastrointestinal tract and ghrelin under experimental
conditions can influence insulin secretion and is reported to increase IA-2
beta but not IA-2 message in islets and inhibition of IA-2 beta with RNAi
decreased ghrelin inhibition of insulin secretion124, 125. Much of the physiological
regulation of insulin secretion can be reproduced in vitro and a wealth of
information has been generated from the study of isolated islets, which appear
to behave as autonomous organs. Such studies have led to the classification of
insulin secretagogues into three groups, initiators, potentiators and
inhibitors. Glucose is the most important initiator in monogastric animals, but
other carbohydrates, amino acids and fatty acids alone can also initiate
insulin secretion. Pharmacological agents like the clinically useful
sulphonylureas tolbutamide and glibenclamide also initiate insulin secretion in
a glucose-independent manner. Arginine initiates insulin secretion by
depolarizing the plasma membrane because of its transport in a positive charge
form. It triggers insulin release by increasing [Ca2+]i 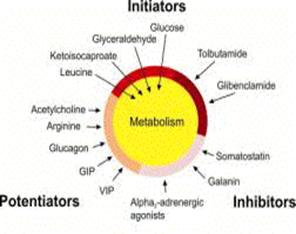 through a KATP channel-independent pathway126, 127. Potentiators of insulin secretion do not initiate insulin secretion by
themselves but enhance stimulation of secretion in the presence of an
initiator. Examples of potentiators are the intestinal glucoincretin hormones
glucagon-like peptide 1 (GLP-1)128 and gastric insulinotropic polypeptide (GIP), which provide fine
regulators of insulin secretion in the post-prandial state along with
neurotransmitters. Other neurotransmitters and hormones such as somatostatin,
galanin, adrenalin and even endocannabinoids (or potentially increase through
CB2 receptors 129 reduce insulin secretion (Fig. 2).
through a KATP channel-independent pathway126, 127. Potentiators of insulin secretion do not initiate insulin secretion by
themselves but enhance stimulation of secretion in the presence of an
initiator. Examples of potentiators are the intestinal glucoincretin hormones
glucagon-like peptide 1 (GLP-1)128 and gastric insulinotropic polypeptide (GIP), which provide fine
regulators of insulin secretion in the post-prandial state along with
neurotransmitters. Other neurotransmitters and hormones such as somatostatin,
galanin, adrenalin and even endocannabinoids (or potentially increase through
CB2 receptors 129 reduce insulin secretion (Fig. 2).
Figure 2. Examples of potentiators, initiators and inhibitors
of insulin secretion from the pancreatic beta cell. Abbreviations used: GIP,
gastric insulinotropic polypeptide; VIP, vasoactive intestinal polypeptide;
GLP-1, glucagon-like peptide 1.
Islet
Pathophysiology in type 1 diabetes
Type 1 diabetes is characterized by the appearance of circulating antibodies
targeted at beta cell proteins, which appear many years before the onset of
clinical disease. Proteins such as insulin, GAD, IA2, IA-2 beta (phogrin) and
CPH (carboxy-peptidase H) are specifically targeted but as yet it is not
settled if any one (or unknown targets) of these molecules is primary or
dominant in the autoimmune response. In the NOD mouse model there is
accumulating evidence that insulin may be a primary autoantigen with knockouts
or alterations of specific insulin sequences preventing diabetes, while
knockouts of GAD65, IA-2, IA-2 beta (and even both IA-2s) do not change the
course of development of NOD diabetes130-132. A recent study of lymphocytes from pancreatic lymph node of patients
with type 1 diabetes also implicated insulin133. Insulin is the only molecule in this group that is specific to the
beta cell though the others have the common feature of being associated with
regulated pathway of secretion. Insulin and CPH are secreted molecules and to
some extent remain on the cell surface following exocytosis. By contrast the
dominant epitopes seen by antibodies reactive to GAD, IA2 and phogrin are
intracellular. It is presently unknown whether the beta cell is targeted
directly or indirectly, whether the antigens are presented by the living cell
or through death or antigen shedding and why the a-cell survives in spite of
sharing many autoantigens with the beta cells and being bathed in the same
milieu of soluble immune effector molecules. One hypothesis is that the beta
cell participates in its own destruction by increased presentation of antigens
in response to glucose, a situation that worsens as beta cell mass decreases
and the remaining cells compensate by increased transcription and translation
of secretory pathway proteins134. Beta and alpha-cells have a differential
sensitivity to cytokines in that inhibition of glucose-stimulated insulin
release by IL-1 is reversible, whereas the effect on glucose-modulated glucagon
release is not135. The beta cells are more sensitive to cytokines than the other three
endocrine cell types in the islets. Cytokine-induced free radicals in beta
cells such as NO catalyzed by the inducible nitric oxide synthase136 may be involved in beta cell-specific destruction in type 1 diabetes137. NO is an important mediator but not the sole mediator of
cytokine-induced cytotoxicity in beta cells as shown by the fact that
inhibition of NO production in human islets did not prevent cytokine-induced
apoptosis, a-cells do not express NO in response to cytokines. Beta cells but
not a-cells induce the expression of heat shock protein 70 (hsp70), heme
oxygenase and MnSOD (manganese superoxide dismutase) upon exposure to IL-1138. Native beta cells have been shown to possess low scavenging potential
for oxygen-derived free radicals and over expression in beta cells of scavenger
or antioxidant enzymes such as MnSOD, catalase and glutathione peroxidase has
been shown to increase their survival after exposure to NO and reactive oxygen
species137. Production of oxygen free radicals mediated by macrophages can damage
beta cells directly resulting in type 1 diabetes in NOD mice139. Superoxide dismutase and catalase protected isolated beta cells
against alloxan-induced diabetes in vivo indicating a role for superoxide
radicals and hydrogen peroxide in the toxicity of alloxan140.
Studies with isolated islets have provided models of how mediators of the
immune response may damage the islet in the autoimmune response in type 1
diabetes. The macrophage cytokine IL-1b induces an initial phase of functional
stimulation in rodent pancreatic islets, which is followed after 4-7 hours by a
progressive inhibition of (glucose-induced) insulin release and eventually
overt damage to the beta cell caused by production of NO136, 141. The two cytokines, TNFa and INFg potentiate the IL-1b-induced
production of toxic NO and oxygen free radicals which inhibits insulin
secretion142, 143. This has led to the concept that IL-1b in combination with INFg and
TNFa plays an important role for beta cell dysfunction and death. The general
process of cytokine-induced beta cell 'de-differentiation' with impairment of
some of the most differentiated functions of beta cells, such as the
preferential mitochondrial metabolism of glucose and the biosynthesis and
release of insulin is paralleled by the activation of proteins related to cell
survival, such as heat shock proteins and antioxidant enzymes144. There are important species differences in beta cell response to IL-1.
IL-1 has stimulatory effects on human islets with no formation of NO compared
to rodent islets, which produces NO when exposed to IL-1. Human islets cultured
in the presence of IL-1b alone exhibit a more prolonged stimulatory phase,
which may last up to 48 hours136. But again, with prolonged exposure of human islets to IL-1b + TNFa +
INFg an inhibition of human islet function is also observed. This difference
between species is probably due to better capacity of human islets to scavenge
oxygen free radicals and to the higher content of hsp70, catalase and superoxide
dismutase in these cells112. Hsp70 has a direct anti-apoptotic role by
inhibiting protein aggregation, decreasing formation of oxygen free radicals
and blocking effector caspases and it could also decrease necrosis by
preventing cellular ATP depletion. The cytokine-induced beta cell
de-differentiation is probably associated to cytokine-induced down-regulation
of pancreas duodenum homeobox gene-1 (PDX-1) and Isl-1. PDX-1 is a crucial gene
for beta cell development and for the maintenance of its differentiated
phenotype (see chapter with Jensen). The decreased expression of PDX-1 together
with the inhibition of Isl-1 expression could contribute to the observed decreased
expression of insulin and glucokinase mRNA which depend on PDX-1 for their
regulated transcription. While IL-1b inhibits PDX-1 and Isl-1 expression and
up-regulate c-myc it does not trigger beta cell apoptosis112.Plasma type II secreted phospholipase A2 (PLA2) may
be closely involved in the pathophysiology of acute pancreatitis. The serum
levels of type II PLA2 has been shown to correlate with the severity of the
disease145. Also, an inhibitor of type II PLA2 protects the pancreas against
tissue damage when pancreatitis is induced in vitro146. Interestingly PLA2 enzyme isoforms are also produced by inflammatory
cells and by the islets themselves (see below). As in the case of the soluble
immune effector molecules we have little information on the actual
concentrations to which the beta cells are exposed and the actual in vivo
response in terms of regulation of receptors and signaling pathways.
The Stimulus-Secretion Coupling of the
Pancreatic beta cell
The pancreatic beta cell is unusual from the biochemical standpoint in that it
has a high rate of glycolytic and oxidative metabolism and that its glucose
metabolism is largely insulin-independent. Most-strikingly it responds to
extracellular glucose concentrations by increased metabolic flux even at
concentrations of the sugar as high as 25 mM. The latter provides a unique
mechanism that couples the availability of metabolizable nutrients to the
control of secretory, translational, and transcriptional processes without the
mediation of ligand-specific cell surface receptors. It may also be the
Achilles heel of the cell in that intracellular proteins are exposed to higher
concentrations of glucose and reactive metabolites than would otherwise occur
in a typical cell that restricts glucose entry and its metabolism to meet its
energetic demands. Within the secretory granule insulin is stored complexes
with zinc and the beta cell has mechanisms to take up zinc147 and transport it into the secretory granule (ZnT-8[expressed only in
beta cells], a cation diffusion facilitator)148. Two other transport systems that are abundant in
islet beta cells and important for islet physiology are GPR40 (G
protein-coupled receptor 40 a free fatty acid receptor) and 4F2 coupled amino
acid transporters. GPR40 mediates the insulin secretory stimulation of fatty
acids such as linoleic acid reportedly through cAMP and protein kinase A
activity, and inhibition of delayed rectifying voltage gated K+ channels149. The 4F2 heavy chain (CD98 heavy chain) is a component of heterodimeric
amino acid transporters that is highly represented on the surface of islet beta
cells150. Of note this molecule is also an
"activation" antigen and present at high levels on malignant cells151, 152.
The beta
cell Glucose Sensor
The beta cell glucose sensor consists of the combination of two molecules,
which have a restricted tissue distribution, namely Glut2 and glucokinase (GK).
Glucose enters the beta cell through the glucose transporter Glut2 (Km ~17 mM)
and is quickly phosphorylated by GK to glucose 6 phosphate (G6P) (Fig. 3)114. Glut2 expression changes in diabetes and hyperglycemic states, which
seems to underline its importance in the response of the beta cell, however
there is great species variation in the expression of this molecule and little
evidence per se that glucose entry is rate limiting to glucose metabolism in
the beta cell. GK on the other hand, although present in other tissues such as
the liver and kidney appears to be driven by a pancreatic specific promoter106, 153, 154. Its high expression and accompanying downregulation of low Km forms of
hexokinases155-157 create a situation where kinetics of the enzyme dictates the kinetics
of the generation of G6P in a cellular context. There has been considerable
debate as to whether the beta cell has a specific glucose 6 phosphatase
(G6Pase) and whether it participates with GK in a regulatory substrate cycling activity158, 159. The beta cell expresses islet G6Pase related protein (IGRP) a beta
cell specific homolog of the liver G6Pase catalytic subunit (55% identity) but
the molecule is apparently not catalytically active160. Of note IGRP is a major target of CD8 T lymphocytes of the NOD mouse133. Other biochemical properties of the beta cell which have been
implicated in its ability to sense metabolic fuels include the absence of
lactate dehydrogenase and thus an inability to re-oxidize cytoplasmically
generated NADH161 and the enhanced expression of the mitochondrial glycerophosphate shuttle162, 163 which may be involved in the transport of reducing equivalents in to
and out of the mitochondria. Anaplerosis (the refilling of Krebs cycle
intermediates) is shown to be implicated in the KATP-independent pathways of
insulin secretion perhaps via malonyl-CoA formation and lipid esterification
processes or through a pyruvate/malate shuttle generating NADPH164. Anaplerosis requires conversion of pyruvate into oxaloacetate by
pyruvate carboxylase an enzyme that is abundant in islet tissue115. Consistent with the view that anaplerosis is an essential component of
beta cell signaling is the fact that the dose dependence of insulin release
highly correlates with the accumulation of citrate, malate, and citrate-derived
malonyl-CoA in the cell164. The fact that the association of rise in citrate and a-ketoglutarate
and initiation of insulin secretion does not require Ca2+ further indicates
that anaplerosis is an early event of beta cell activation131. A net result of this biochemical machinery is that rates of glucose
metabolism are strongly correlated to changes in the redox state of pyridine
nucleotides (particularly NADPH165, 166 and the ratio of ATP/ADP in the cell both which may act as
intracellular signals for ionic and other events in the cell (Fig. 3)167. The beta cell is electrically excitable and when
stimulated with glucose it shows oscillations in membrane potential
characterized by bursts of 5-15 sec duration. Increasing glucose lengthen the
burst and shortens the interval leading to continuous spiking over a base of DY
<-40 mV168, 169. The beta cell resting membrane potential of about -70 mV is mainly
determined by the activity of ATP-sensitive K+ channels (KATP)66. When the ATP/ADP ratio increases as a result of
glucose metabolism, the KATP channels in the plasma membrane close170, 170-176. This causes the cell membrane to depolarize due to the presence of an 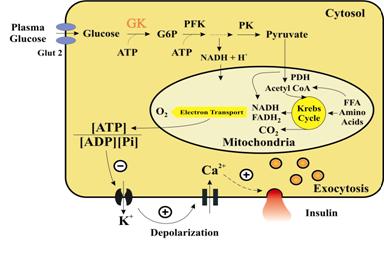 inwardly
directed cation current that is dominant in the absence of a reduced
KATP-conductance. The nature of this current is currently unknown but may be a
member of the trp family that carries Na+ and Ca2+ ions177. The resulting membrane depolarization leads to opening of the
voltage-dependent L-type Ca2+ channels in the plasma membrane. The resultant
Ca2+ influx elevates the intracellular free Ca2+ concentration [Ca2+]i and
initiates exocytosis of insulin-containing secretory granules (Fig. 3).
inwardly
directed cation current that is dominant in the absence of a reduced
KATP-conductance. The nature of this current is currently unknown but may be a
member of the trp family that carries Na+ and Ca2+ ions177. The resulting membrane depolarization leads to opening of the
voltage-dependent L-type Ca2+ channels in the plasma membrane. The resultant
Ca2+ influx elevates the intracellular free Ca2+ concentration [Ca2+]i and
initiates exocytosis of insulin-containing secretory granules (Fig. 3).
Figure 3.
Stimulus-secretion coupling in the pancreatic beta cell. Glucose is transported
across the plasma membrane through the GLUT2 transporter. Glucose is metabolized
through the glycolysis and Krebs (TCA) cycle. The increased ATP/ADP ratio leads
to closure of the ATP-sensitive K+ channel in the plasma membrane, membrane
depolarization and opening of the voltage-dependent Ca2+ channels. The
resulting Ca2+ influx stimulates release of the insulin-containing granules by
exocytosis. [Note: Trp is the transient receptor potential gene family that
first was cloned from Drosophila where it function in photoreception, but which
seems to include Ca2+ store-operated channels and inositol
1,4,5-trisphosphate-activated channels.]
Besides the
effect of glucose on the KATP channels glucose is also shown to affect the
stimulus-secretion coupling independently of the KATP channels thereby
amplifying the pathway of glucose-induced insulin secretion178. This has been shown by the fact that glucose dose-dependently
increases insulin secretion when the cell is depolarized and KATP channels
cannot be closed (diazoxide plus high K+)179. Under conditions when the KATP channels are closed by sulfonylureas
and therefore the beta cell plasma membrane is depolarized and insulin
secretion is increased, glucose is shown to be able to further amplify insulin
secretion dose-dependently. This KATP channel-independent (amplifying) effect
of glucose on insulin secretion is Ca2+-dependent but not mediated by any
further rise in [Ca2+]i179. The amplifying pathway serves to augment the secretory response
induced by the triggering signal and creates dose-dependent secretory response
in the 5-25 mM concentration range. Glucose may also regulate insulin secretion
by a Ca2+-independent mechanism180-182. Under stringent Ca2+-deprived conditions when protein kinases A and C
are activated simultaneously, glucose stimulates insulin release in the absence
of an elevation of [Ca2+]i. This Ca2+-independent amplification of glucose-induced
insulin release may require GTP late in stimulus-secretion coupling183, 184. This has been interpreted in terms of two different mechanism of
exocytosis, one driven by Ca2+ and the other by GTP. Others argue that
glucose-induced insulin release is mainly regulated by the two Ca2+-dependent
pathways (rise in beta cell [Ca2+]i and increase in Ca2+ efficacy) without
significant contribution of a Ca2+-independent mechanism. In the following,
some of the key molecular components in the beta cell stimulus-secretion
coupling will be described.
The
ATP-Sensitive K+ Channel Complex
KATP channels are expressed in different cell types including islets, heart,
muscles and ventromedial hypothalamus (VMH) and serve to couple cell metabolism
to membrane excitability. They are composed of a pore-forming complex
consisting of subunits, a specific K+ channel (Kir6.2) surrounded by regulatory
sulphonylurea (SUR) binding subunits. See reviews185-187. SUR (Mw 140 kDa) is a member of the ABC-transporter superfamily and
Kir6.2 (390 amino acids, 35 kDa) a member of the inward rectifier K+ channel
superfamily. Four subunits of Kir6.2 and SUR1 each constitute the functional
channel complex in islets188, 189. KATP channel activity is modulated by a range of compounds and
intracellular metabolites. Channel activity is reduced by Mg-ATP and
sulphonylureas, the most commonly used class of compounds in the treatment of
type 2 diabetes. ADP and diazoxide conversely activate KATP channel activity.
Changes in the beta cell ATP/ADP ratio are an important physiological regulator
of channel activity with an increase in the blood glucose concentration leading
to an enhanced ATP/ADP ratio and consequently channel closure. Recently, KATP
channels have also been detected in a- and d-cells189-191, whereas the functional consequence of channel closure in the d-cell is
similar to that in the beta cell (increased hormone release), inhibition of
channel activity in the a-cell leads to inhibition of glucagon release192. The physiologic importance of this channel in man has become abundantly
clear in the past several years with the discovery that approximately ½ of
neonatal diabetes results from activating mutations of the Kir6.2 molecule.
There is a hierarchy of disease dependent on the severity of the functional
effect of the mutations leading to transient neonatal diabetes,
permanent neonatal diabetes, and finally a syndrome with mental retardation and
neonatal diabetes (DEND syndrome: developmental delay, epilepsy, and neonatal
diabetes193-195. Paternal inactivating mutations of the genes for Kir6.2 and SUR1
combined with loss of the corresponding maternal chromosomal (11p15) region in
focal areas of the pancreas leads to focal islet lesions, while autosomal
inheritance of mutations results in diffuse pancreatic lesions, and both cause
hyperinsulinemic hypoglycemia185, 196-201. Currently the genetic bassis of congential hyperinsulinemia have been
recognized due to associations and mutations in eight genes: ABCC8, KCNJ11,
HADH1, GCK, GLUD1, SLC16A1, UPC2 and HNF4a202-205.
|
HYPERINSULINEMIA GENES
|
|
GENE
|
PROTEIN
|
LOCUS
|
INHERITANCE
|
|
ABCC8
|
SUR1
|
11p15.1
|
Recessive
/Dominant
|
|
KCNJ11
|
Kir6.2
|
11p15.1
|
Recessive /Dominant
|
|
HADH1
|
SCHAD
|
4q22-q26
|
Recessive
|
|
GK
|
GK
|
7p15-p13
|
Dominant
|
|
GLUD1
|
GDH
|
10q23.3
|
Dominant
|
|
SLC16A1
|
MCT1
|
1p13.2-p12
|
Dominant
|
|
UCP2
|
UCP2
|
11q13
|
Dominant
|
|
HNF4a
|
HNF4a
|
20q12-q13.1
|
Dominant
|
Mutations in HADH1 which encodes fatty
acid oxidation enzyme SCHAD causes
recessive hyperinsulinemia206. Dominant mutations of GCK cause hyperinsulinemia that is resistant to
therapy207, 208. Dominant mutations of GLUD1 which encodes mitochondrial glutamate
dehydrogenase is characterized by hyperammonemia209. It is thought that glutamate dehydrogenase mutations lead to increased
ATP in islet beta cells, closing the K sensitive ATP channel and thus
inappropriate insulin secretion and hypoglycemia. Abnormalities of mitochondria
are increasingly recognized as central to beta cell abnormalities and in
particular there are a series of mitochondrial mutations that lead to diabetes
and more recently reports that mitochondrial "stress" and
abnormalities related to mitochondrial "uncoupling" are important for
insulin secretion defects in type 2 diabetes210, 211.
The Beta Cell Calcium Channels
Membrane depolarization, resulting from KATP channel closure leads to elevation
of cytosolic Ca2+ via voltage-gated Ca2+ channels in the plasma. The influx of
Ca2+ in turn regulates several steps in exocytosis, such as the size of the
readily releasable vesicle pool, the fusion event, and expansion of the fusion
pore212. The Ca2+ concentration required for initiating insulin secretion in
different experimental systems has been estimated in the range of 10-30 mM213, 214. The resting level is in the submicromolar range.
Pancreatic beta cells are found to express N-, P/Q- and L-type Ca2+ channels,
with the major contribution to Ca2+ influx mediated by the L-type channel
(large and long-lasting)66. The L-type channels are characterized by the
requirement of a strong depolarization to activate. They inactivate slowly,
which is seen in a patch-clamp experiment during a voltage-clamp depolarization
to 0 mV where channel inactivation is maximal (Fig. 4)215. The L-type channels are by themselves inactivated
by Ca2+ ions at the inner side of the membrane, providing a negative feedback
mechanism that limits Ca2+ entry into the cell216. There is also a voltage-dependent component in the
inactivation The beta cell L-type Ca2+ channel activity is blocked by
dihydropyridines e.g. nifedipine and stimulated by the cyclic AMP/protein
kinase A pathway. Elevated cAMP levels lead to a reduced rate of Ca2+ channel
inactivation whereas the peak current is only moderately increased217.
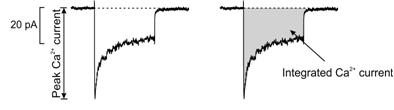
Figure 4. Ca2+ current in a mouse beta cell is mediated by
L-type Ca2+ channels. The peak amplitude and the integrated Ca2+ current are
depicted resulting from a membrane depolarization to 0 mV during a
voltage-clamp experiment.
Molecular Motors and SNARES
When the Ca2+ channels in the plasma membrane open and Ca2+ flows in the cell,
the onset of exocytosis follows less than 50 ms later. This latency is shorter
than the time required for Ca2+ to equilibrate in the cytosol, which suggests
that the granules are located in the vicinity of the Ca2+ channels and thus
sensitive to local [Ca2+]i changes. Indeed, areas with a high density of Ca2+
channels are found to co-localize with the granules and represent 'hot spots'
of secretion158. A mouse beta cell contains about 13,000 secretory
granules218 but only a small fraction (50-75) granules219 in the readily releasable pool (RRP) are available for immediately
release220, 221. The remainder referred to as the reserve pool needs to be mobilized
into the RRP before they can undergo exocytosis. The number of granules
released seems to be dependent on the phosphorylation state of key proteins.
Activation of protein kinase A and C (PKA and PKC) or inhibition of protein
phosphatases is required for a large exocytotic response217. The effects of these kinases are additive, which suggests that
phosphorylation of different regulatory proteins are involved.
Glucose-stimulated insulin secretion in vivo is typically biphasic and
characterized by a steep transient first phase is followed by a gradually
developing second phase (Fig. 5). In one model it is explained as the first
phase represents release of granules in RRP, triggered by Ca2+ influx, and the
second phase most likely reflects the ATP-dependent recruitment and release of
granules from the reserve pool221. The electrophysiological response is also biphasic
so this conclusion is open to debate.
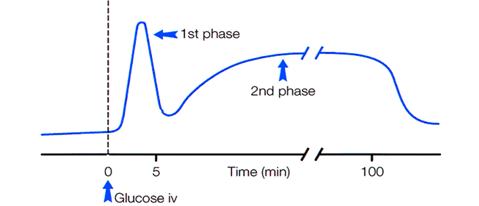
Figure 5.
Glucose-induced insulin secretion is characterized by a rapid first phase and a
slower second phase insulin released to the bloodstream. Abbreviation used: iv,
intra venous.
 Mobilization
of granules may involve physical translocation within the beta cell and/or
chemical modification222 and docking of granules near the plasma membrane. Docked granules need
to be primed in order to enter the RRP. The insulin granules are transported
from the reserve pool to the plasma membrane initially along microtubules and
then along the microfilament networks of the cytoskeleton associated with the
plasma membrane223. An ATP-dependent motor protein such as kinesin is thought to provide
the necessary motive force for transportation. However, the exact mechanism and
its regulation involved in the transport of granules towards the plasma
membrane remains largely unknown224.
Mobilization
of granules may involve physical translocation within the beta cell and/or
chemical modification222 and docking of granules near the plasma membrane. Docked granules need
to be primed in order to enter the RRP. The insulin granules are transported
from the reserve pool to the plasma membrane initially along microtubules and
then along the microfilament networks of the cytoskeleton associated with the
plasma membrane223. An ATP-dependent motor protein such as kinesin is thought to provide
the necessary motive force for transportation. However, the exact mechanism and
its regulation involved in the transport of granules towards the plasma
membrane remains largely unknown224.
The processes by which granules in close proximity to the plasma membrane bind
and fuse to it can be divided into three phases docking, priming and fusion
based on electrophysiological and secretion studies performed on various neurosecretory
tissues and in combination with biochemical and molecular genetic analysis.
Docking describes the initial reversible interaction of granules with the
plasma membrane and constitutes a pool of unprimed granules in close proximity
to the exocytotic site. There is little morphological evidence of the existence
of granule docking, but the presence of this pool in the beta cell is suggested
from the speed by which RRP is replenished following addition of ATP in the
cytosol. The next priming step may involve ATP-hydrolysis mediated in part by
the ATPase N-ethylmaleimide-sensitive factor (NSF), which is activated by the
soluble NSF-attachment factor (a-SNAP)225. NSF functions as a molecular chaperone to activate SNAp REceptor
(SNARE) proteins destined for the fusion complex (Fig. 6)224. Another possible ATP-dependent event in priming is
the synthesis of phosphatidylinositol (4,5)-bisphosphate (PIP2), which is
thought to be important in the recruitment and modulation of proteins
implicated in the fusion apparatus - namely the Ca2+ binding synaptotagmin and
calcium-dependent activator protein for secretion (CAPS)212, 226. Multiple different molecules influence insulin granule exocytosis. The
transcription factor HNF-1 when mutated is a cause of a severe form of MODY
(Maturity Onset Diabetes of Youth) and in addition to its many effects upon
islets and insulin secretrion, the molecule collectrin is regulated downstream
of HNF-1, and collectrin under or over expression leads to decreased or
enhanced insulin secretion227. Insulin secretion is deficient in a mouse with a mutant Akt/PKB kinase
and this protein regulates exocytosis in a model system228.
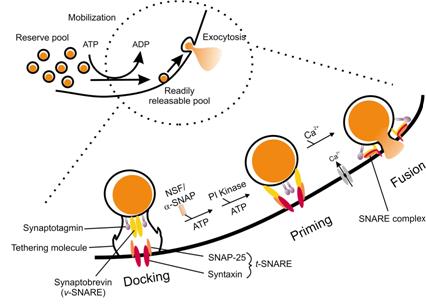
Figure 6.
Docking, priming and fusion of insulin-containing granules. Granules from the
reserve pool needs to be mobilized in order to enter RRP prior to release. The
insert illustrates the three primary stages of insulin exocytosis at the plasma
membrane, docking, priming and fusion. The granule is tethered at the plasma
membrane during docking. Thereafter the a-SNAP activates NSF, which hydrolyses
ATP thereby activating the SNARE proteins. A second ATP-dependent step in the
priming process could be PI-4kinase phosphorylation in the generation of
PIP2 (after PI-5kinase phosphorylation). Synaptotagmin is proposed to act as a
Ca2+ sensor and a fusion clamp preventing fusion until a Ca2+ signal arises. When the
granule is primed, Ca2+ is the only requirement for granule fusion with the
plasma membrane.
Fusion of granules
Conserved protein
families involved in fusion include the SNAREs, the Rab GTPases, and the
Sec1/munc18-related proteins (also referred to as SM proteins)229. Granuphilin is reported to be in dense core
vesicles of beta cells and binds to GTP-bound Rab27a230. Rab27a, on the granule membrane is involved in the
regulation of exocytosis of secretory granules. Granuphilin also directly
interacts with plasma membrane bound SNARE protens230. Insulin granules docked to the membrane are reduced
in granuphilin deficient beta cells231 despite granuphilin null mice having augmented insulin secretion231. The SNARE proteins were originally identified as synaptic proteins,
but are now generally accepted as universally involved in the core machinery in
all intracellular fusion events. The primed granules are ready to fuse with the
plasma membrane in an ATP-independent process when the intracellular Ca2+
concentration [Ca2+]i am sufficiently elevated.
Synaptobrevin (VAMP-2, synaptic vesicle-associated membrane protein) is located
on the beta cell granule as a v-SNARE, and syntaxin 1 and SNAP-25
(synaptosome-associated protein of 25 kDa) are located on the plasma membrane
as t-SNARE (Fig. 6)232, 233. Synaptobrevin, SNAP-25 and syntaxin 1 assemble in a 1:1:1
stoichiometry to form a complex that drives membrane fusion. Helical portions
in the SNARE complex bundle together (two helical domains from SNAP-25 and one
from both syntaxin 1 and synaptobrevin) and form an extended coiled-coil
rod-like structure (see note), which brings the granule into close proximity
with the plasma membrane, thereby overcoming the free energy barrier for fusion
(Fig. 6)234.
[Note: Coiled-coil structure is composed of several a-helices that interact
through hydrophobic residues and stabilizing electrostatic interactions between
the side chains. This results in a very stable structure resembling a strong
rope.]It has been postulated that munc18 functions in a late stage in the
fusion process in chromaffin cells, where its dissociation from syntaxin 1
determines the kinetics of postfusion events235. However, a munc18 null
mutation leads to a selective defect in the docking of LDCVs at the target
membrane and overexpression of munc18
leads to an increase in number of fusion-competent vesicles without affecting
the kinetics of vesicle fusion. This suggests that munc18 is also important in
the docking step before trans-SNARE complexes have assembled. The essential
role of these proteins in neurosecretion is illustrated by the potent
inhibition of insulin secretion by various botulinum and tetanus toxins that
specifically cleave the different SNARE proteins. Studies with botulinum
neurotoxin A which cleaves VAMP-2 and SNAP-25 demonstrate that VAMP-2 is
absolutely necessary for insulin exocytosis whereas the action of SNAP-25 could
be partially reversed by higher levels of Ca2+ or cAMP potentiation236. Botulinum neurotoxin C1 mainly cleaves syntaxin and reduces K+-induced
insulin release by 95% but glucose stimulated insulin release only by 25%
pointing to further complexity in the exocytotic mechanism237. The fusion event is highly Ca2+-dependent (>10 mM), probably due to
regulatory proteins that function as Ca2+ sensors. Synaptotagmin is the
best-characterized exocytotic Ca2+ sensor with two Ca2+-binding regions, the C2
domains (see note) C2A and C2B, each sharing homology with the C2 regulatory
domain of protein kinase C (PKC). The C2A domain is responsible for the
Ca2+-dependent insertion of synaptotagmin into the beta cell granule membrane
at low micromolar (5 mM) free Ca2+ and the binding of synaptotagmin to the core
complex protein syntaxin 1224, 238. CAPS is another Ca2+-binding protein thought to be important for
Ca2+-triggered fusion from neuroendocrine cells, including the beta cell224. It contains a pleckstrin homology domain (see note)
that exhibits binding specificity towards PIP2, which makes it a specific
target for phospholipid mediators formed during priming. The lipid specificity
of CAPS is switched at elevated Ca2+ concentrations and is thought to help
disrupt the plasma membrane bilayer to permit fusion239. Synaptotagmin is also found to have an inhibitory function in
exocytosis. It has been suggested to lock the fusion core complex by preventing
a-SNAP from binding until a stimulatory Ca2+ signal arises225, 233. Other components of the fusion process include syntaphilin that acts
as a syntaxin clamp in regulating assembly of the SNARE complex during membrane
fusion events234 and NSF that may be necessary to dissociate SNARE complexes formed
within a single membrane in favor of complexes between membranes (trans-SNARE
complex)240, 241. [Note: A C2 domain contains about 120-140 amino acids and has been
found in single or multiple copies in more than 60 proteins. It was first
discovered in PKC as a Ca2+-dependent, phospholipid-binding domain, but now
several other C2 domains have been shown to mediate protein-protein
interactions and Ca2+-independent binding to proteins and phospholipids242, 243. Pleckstrin homology domain mediates membrane association of kinases
with their target molecules, in some cases by interaction with the headgroup of
a phosphoinositide lipid243.
Rab GTPases represent a large family of homologous Ras-like GTP-binding
proteins that direct the vectorial movement of secretory vesicles. Four
isoforms of Rab3 (Rab3A, -B, -C, and -D) have been identified so far. Rab3A is
associated with dense-core insulin-containing secretory granules and thought to
play a negative role in the fusion process by preventing Ca2+-dependent
exocytosis244. Rab3A activation is shown to inhibit Ca2+-evoked exocytosis in beta
cells by possibly binding to calmodulin at low stimulatory [Ca2+]i245. Calmodulin is a key Ca2+ sensing protein and this may be a way for
Ca2+ to act in concert with GTP in the insulin exocytotic mechanism (provides a
downstream connection between an intracellular secondary messenger (Ca2+) and
the exocytotic machinery of the pancreatic beta cell). It is postulated 246 that it is a way to direct calmodulin to the
cytoplasmic face of a b-granule membrane. When [Ca2+]i increases, calmodulin
binds CaMKII more readily than Rab3A and this activates CaMKII to phosphorylate
effector exocytotic molecules and enhance either the transport of a b-granule
to the plasma membrane and/or the mechanism of insulin exocytosis187.
Ca2+ plays another essential role in triggering endocytosis following
secretion. This may be controlled by the Ca2+/calmodulin-dependent
serine/threonine protein phosphatase calcineurin, which is postulated to be a
calcium sensor for endocytosis in synaptosomes. Calcineurin is activated by
calcium and thereby leads to dephosphorylation of proteins involved in
endocytosis like dynamin, amphiphysin 1, amphiphysin 2, and synaptojanin247, 248.
Signaling inside the beta cell
Metabolizable secretagogues and
extracellular molecules such as hormones, neurotransmitter that act by binding
to receptors on the beta cell plasma membrane appear to mediate their effects
through a common set of second messengers. These effects include 1) modulation
of adenylate cyclase-cAMP system and activation of protein kinase A (PKA) and
recent evidence that cAMP may influence the ATP-sensitive Potassium channel by
signaling through Epacs (Exchange Proteins Activated by Cyclic AMP)249, 2) generation of inositol 1,4,5-triphosphate (IP3)
and sn-1,2-diacylglycerol (DAG) resulting in activation of the PKC and finally
3) changes in intracellular free Ca2+ ions that function through Ca2+-dependent
regulatory proteins such as calmodulin and PKC. Phosphoinositide signaling
appears to also be important for insulin exocytosis with recent evidence using
RNAi technology of effect through phospholipase D1, Ca2+ dependent activator
protein for secretion 1, and Munc-18-interacting protein 1250.
Hormones binding to G-protein coupled receptors on the cell surface signal to a
heterotrimeric seven transmembrane G-protein (inhibitory G-protein, Gi or
stimulatory G-protein, Gs), which convey the signal to effector systems such as
adenylate cyclase. When a stimulatory G-protein is activated (e.g. glucagon
stimulation of liver cells) the adenylate cyclase, which resides on the inner
plasma membrane, converts ATP to the second messenger cyclic AMP (cAMP) that
activates the cAMP-dependent serine/threonine kinase PKA. PKA phosphorylates
several proteins at their serine or threonine residues causing a change in
either enzyme activity or protein structure. An example is the activation of glycogen
mobilization by activating glycogen phosphorylase.
cAMP has a short half-life in the cytoplasm and it is quickly hydrolyzed to AMP
by the action of cAMP phosphodiesterases which is important for shutting down
the stimulation by the second messenger. cAMP also potentiates Ca2+-dependent
insulin secretion251-253. Besides acting on the Ca2+ channels, the major action of cAMP on
insulin release is by a direct effect on the secretory machinery itself251, 254. This is thought to be a result of an increased Ca2+ sensitivity of
exocytosis by cAMP via an extension of the distance over which the Ca2+
channels can recruit the secretory granules for release251. It has been suggested that cAMP acts by increasing
the release probability of granules already in RRP (PKA-independent) and by
accelerating granule mobilization thereby refilling the RRP (PKA-dependent)253. A recent study has reported that Epac-selective cAMP analogs, but not
PKA-selective analogs influence K ATP channels. Epac 1 and 2 bind to SUR1 of
the ATP K+ channel, and thus cAMP may stimulate insulin secretion through a
novel Epac mediated mechanism249. Other G-protein receptors (inhibitory and stimulatory G, Go;
stimulatory G, Gq) are coupled to phospholipase C (PLC). Acetylcholine
stimulates exocytosis by activating the cholinergic muscarinic receptor that
acts via a stimulating G-protein (Gq), which is bound to the Ca2+-dependent
membrane-bound PLC. PLC hydrolyses PIP2 in the membrane to yield the two second
messengers IP3 and DAG. IP3 diffuses to the endoplasmic reticulum (ER) where it
binds to and opens a Ca2+ channel in the ER membrane thereby allowing Ca2+ to
exit from the ER stores into the cytosol resulting in increased [Ca2+]i. The
membrane bound DAG stimulates protein kinase C (PKC) by lowering its
requirement for Ca2+. DAG is also produced when PLC is activated following
increases in [Ca2+]i, thus PKC will be activated under all conditions in which
intracellular Ca2+ is elevated254. PKC is shown to modulate Ca2+-dependent insulin secretion217, 251. However, glucose-stimulated insulin secretion is unaffected in
PKC-depleted islets, indicating that this enzyme is not essential for glucose
stimulated insulin secretion. Rather, PKC appears to be involved in the
stimulation of insulin secretion by potentiators of release e.g. acetylcholine
and carbamylcholine by sensitizing the secretory machinery to Ca2+217. Glucose-induced insulin secretion is associated with inhibition of
free fatty acid (FFA) oxidation, increased esterification and complex lipid
formation by pancreatic beta cells. Increases in the mass of DAG, triglyceride
and phosphatidic acid are observed. Exogenous FFA also potentiate
glucose-stimulated secretion of insulin, possibly by providing additional acyl
groups for long chain-CoA formation or complex lipid synthesis. FFA does not
stimulate insulin secretion in the absence of glucose probably due to their
rapid entry into the mitochondria when malonyl-CoA levels are low. 116 LC-CoA
opens the ATP-sensitive K+ channel whereas the corresponding FFA closes the
channel. Long-chain fatty acids stimulate PKC, but the saturated ones are
ineffective 254 or much less effective than the unsaturated ones255.
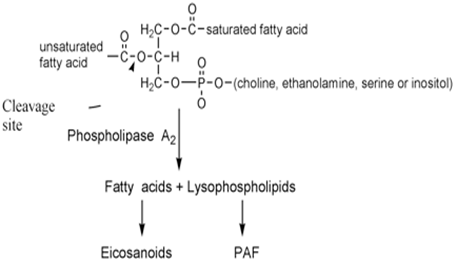 Phospholipase A2
Superfamily and Pancreatic beta cell Function
Phospholipase A2
Superfamily and Pancreatic beta cell Function
The phospholipase A2 family plays an important role in the inflammatory
response seen in IDDM by generating inflammatory molecules from lipids
substrate. Phospholipase A2 (PLA2) constitutes a growing superfamily of enzymes
that hydrolyze membrane phospholipids at the sn-2 position generating lipid
second messengers as well as structural modulations of cellular membranes (Fig.
7). The fatty acid released from sn-2 is in most cases arachidonic acid.
Figure 7.
Illustration of cleavage sites for phospholipase A2 in the glycerol backbone of
a phospholipid. Phospholipase A2 cuts the phospholipids in the sn-2 position
resulting in formation of a fatty acid and a lysophospholipid, which can be
further metabolized to eicosanoids and PAF, respectively. Abbreviations used:
PAF, platelet-activating factor (modified from (171).
Several different
types of PLA2 have been identified. They include: 1) cytosolic PLA2 (cPLA2), 2)
secreted PLA2 (sPLA2), 3) Ca2+-independent PLA2 (iPLA2) and 4)
platelet-activating factor acetylhydrolase (PAF-AH). These groups are further
subdivided according to structure, molecular weight, substrate specificity,
cellular localization, and Ca2+ dependency of the enzyme256. The cytosolic forms of PLA2 (group IV PLA2) are
high molecular weight interfacial PLA2 enzymes (see note). They show a
preference for phosphatidylcholine (PC) with arachidonic acid (AA) in the sn-2
position and are thought to play an important role in providing free AA for
eicosaniod biosynthesis242, 243, 257. Type IV knock-out mice (IV cPLA2a-/- mice) show deficient inflammatory
response 258 and peritoneal macrophages from IV cPLA2a-/- mice fail to produce
prostaglandins and leucotrienes after stimulation259. [Note: Interfacial PLA2 enzymes need to bind to an aggregated surface
in order to access the phospholipid substrates.]
Group I, IIs (IIA, IIC, IID, IIE, IIF), III, V, IX, X and XI PLA2 comprise a
group of low molecular weight PLA2 (13-18 kDa) interfacial enzymes which are either
secreted or extracellular PLA2 (sPLA2). These enzymes have been implicated in
various physiological and pathological functions, including lipid digestion,
lipid mediator generation, cell proliferation, exocytosis, antibacterial
defense, inflammatory diseases and cancer260.
The Ca2+-independent PLA2 (iPLA2) is a group of cytosolic PLA2 of 85-88 kDa
whose major cellular function is the mediation of phospholipid remodeling and
homeostasis246, 261, 262. This enzyme seems to regulate the incorporation of AA and other fatty
acids into membrane phospholipids and as such may be a limiting factor for
eicosanoid biosynthesis though it has been questioned in the case of the rat
pancreatic islet group VIA enzyme263.
Platelet-activating factor acetylhydrolase (PAF-AH) is another group of
Ca2+-independent PLA2's264. All of them have restricted substrate specificity
and are selective for PAF (see note) and for phospholipids with a short (or
intermediate) acyl chain at the sn-2 position that protects normal membrane
lipids from hydrolysis by PAF-AH. A second group of substrates is oxidatively
fragmented phospholipids. The main function of PAF-AH is to act as scavenger of
bioactive phospholipids especially to regulate the level of PAF265. (Note: PAF
(1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine), a bioactive phospholipid
involved in asthma. It is a mediator of a wide range of immune and allergic
reactions. It is synthesized in a regulated pathway and activates inflammatory cells.
Lysophospholipid can be converted to PAF.)
Different types of PLA2 in islets and
pancreatic beta cells
Pancreatic islets contain at least five different types of PLA2 (Table 2).
Among the secreted form, both type IB and IIA are found with type IB more
abundant. Type IB has been localized to the secretory granules of beta cells
and stimulation with insulin secretagogues results in the co-secretion of
insulin and the enzyme263. Type IB also appears to be regulated by fasting and ambient glucose263, 266, 267. Furthermore, mRNA for a sPLA2 membrane receptor is found in rat
pancreatic islets.

Table 1. PLA2 groups in the
pancreatic islet.
By use of
different inhibitors of PLA2, it has been demonstrated that Ca2+-dependent PLA2
may participate in acetylcholine-induced activation of the granule movement in
pancreatic beta cells268. It is proposed that the type IV cPLA2 couples the secretagogue induced
increase in intracellular Ca2+ to the liberation of AA and the subsequent
release of insulin269. Both exogenous sPLA2 and products of its action have also been shown
to suppress KATP channel activity in excised and cell-attached patches of
plasma membranes from HIT insulinoma cells270.
In glucose-stimulated islets, hydrolysis of AA from membrane phospholipids
appears in part to be mediated by a PLA2 that is catalytically active in the
absence of Ca2+ and inactivated by the iPLA2 inhibitor HELSS (haloenollactone
suicide substrate)224. However, iPLA2 is not required for AA incorporation
or phospholipid remodeling in beta cells, suggesting that iPLA2 plays another
role in these cells. iPLA2 in rat and human beta cells is implicated as a
putative glucose sensor, which can couple alterations in glycolytic metabolism
to the generation of biologically active eicosanoids and thereby facilitate
glucose-induced insulin secretion271. iPLA2 is activated by ATP and other nucleotides and it has been
postulated that it may respond to the increase in the ATP/ADP ratio after
glucose stimulation, however this function has been questioned.
Besides playing an important role in the stimulus-secretion coupling in the
beta cell different types of PLA2 may be involved in inflammatory responses.
Type II sPLA2 and type IV cPLA2 mRNA is upregulated in areas with histologic
damage in pancreatitis suggesting that these isoforms might contribute to the
morphologic changes that occur272. In addition, several proinflammatory cytokines such as IL-1, IL-6 and
TNFa have been shown to induce transcription of the type IIA sPLA2 gene in a
variety of cells. TNF priming of human neutrophils (PMN) caused marked increase
in fMLP stimulated AA release in parallel to enhanced activity and secretion of
sPLA2 and activity of cPLA2273. Type I sPLA2, on the contrary, is non-toxic to acinar cells in both
severe and mild pancreatitis. This may indicate that type II but not type I
sPLA2 have pathophysiologic importance in severe acute pancreatitis associated
with systemic inflammatory response syndrome.
Arachidonic
acid in cell signaling
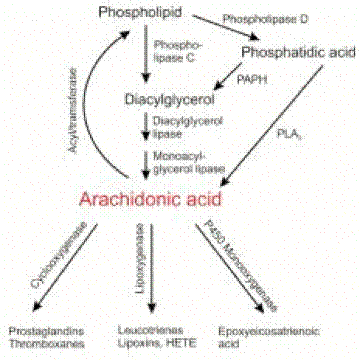
Arachidonic acid is a major constituent of islet phospholipids comprising about
34-39% of the sn-2 fatty acyl mass in islet glycerophospholipids 271, 274, 275 compared to 13% in rat brain and heart and 1% in rat liver276. The intracellular level of free AA in islets is normally in the low
micromolar range, but rises to a level of 50-200 mM during glucose stimulation277, 278. Such changes could result by the action of PLA2, but also by the
action of diacylglycerol lipase and monoacylglycerol lipase on diacylglycerol
(DAG), generated from the action of phospholipase C (Fig. 10)279, 280. Phosphatidic acid (PA), generated by phospholipase D hydrolysis of PL,
is also a potential source of AA279.
Figure 8.
Arachidonic acid can be made in different ways, but also degraded in several
ways. AA can be reesterified into membranes or metabolized by the lipoxygenase
pathway to leucotrienes, by the cyclooxygenase pathway to prostaglandins and
thromboxanes or by the cytochrome P450 monooxygenase enzyme to
epoxyeicosatrienoic acid. Abbreviations used: PAPH, phosphatidic acid
phosphohydrolase.
Inhibition of AA
release prevents insulin secretion281. Increases in the availability of AA (or its metabolites) by itself,
however is probably insufficient to initiate insulin release, instead it seems
that AA potentiates the action of other secretagogue282. In different cell types, AA has been associated
with several specific cellular processes such as regulation of PKC and PLC and
modulation of intracellular Ca2+ transients283-286. In beta cells, it has been suggested that both Ca2+-dependent and
Ca2+-independent components are involved in PLA2-induced insulin secretion. The
Ca2+-independent component could be a result of AA or a lipoxygenase product
affecting late events in stimulus-secretion coupling287, 288. The AA-induced Ca2+-dependent event is biphasic with an early rise
probably reflecting release of Ca2+ from intracellular stores and a sustained
phase, which could reflect extracellular Ca2+ entry possible mediated by
voltage-sensitive Ca2+ channels 289
Role of arachidonic acid metabolites in
cell signaling
When AA is generated, it can be reesterified into phospholipids by
acyltransferases or metabolized by the cyclooxygenase (COX), lipoxygenase (LPX)
or cytochrome P450 epoxygenase pathways generating prostaglandins and
thromboxanes, leukotrienes and lipoxins, or epoxyeicosatrienoic acids290. Prostaglandins made by the
cyclooxygenase-pathway acting on AA have been demonstrated, in cell-attached
patches on a beta cell line, to be involved in insulin secretion by increasing
KATP channel activity270. E Prostaglandins inhibit insulin secretion and a pertussis toxin
insensitive G alpha i family member G alpha z apparently mediates this effect291. The lipoxygenase metabolites of AA are potential mediators of
insulin secretion. During nutrient-induced insulin secretion
12-hydroxyeicosatetraenoic acid (12-HETE; a product of 12-lipoxygenase
metabolized AA) is augmented282 and a lipoxygenase inhibitor (nordihydroguaiaretic
acid, NDGA) inhibits glucose-induced insulin secretion292. Products of the lipoxygenase pathway have also been
demonstrated to play a role in apoptosis induced by Ca2+ store depletion in
beta cells by using the inhibitor NDGA293.
The cytokine IL-1 also augments islet production of 12-HETE and PGE2276. This is not caused by an increased production of AA but by suppression
of reesterification of AA into islet phospholipids. This might occur as a
result the IL-1-induced production of NO and inhibition of ATP-dependent
conversion of the fatty acid to a coenzyme A thioester intermediate276. 12-HPETE, a precursor of 12-HETE, has been shown to induce apoptosis
of cells with reduced levels of glutathione peroxidase. Thus the effects of
IL-1 to increase substrate flux through the lipoxygenase pathway and to augment
NO production may interact cooperatively to inflict injury on beta cells, and
at the same time stimulate insulin release in the short term.
Pathophysiogical role of lipid
metabolites and lysophospholipids on beta cell signaling
Inflammatory
response in the pancreas not only plays an important role in the destruction of
islet tissue in autoimmune diabetes (see chapter by Mandrup Poulsen), but also
in the ability of the pancreatic beta cell to recover from such insult. Pancreatitis
induced by cellophane wrapping (CW), surgery, coxsackievirus B4, cerulein
treatment or with other chemicals294-296 for example can lead to islet regeneration. Locally produced INFg
causes lymphocyte infiltration and islet cell destruction and while IFN-g gene
linked to an insulin promoter produces diabetes new cells are formed from duct
epithelial cells and they differentiate into endocrine cells297. The proinflammatory cytokines, IL-1, TNF and perhaps IL-6 is shown to
induce PLA2 expression and release298. The proinflammatory action of TNF depends in part
on activation of PLA2299. In humans with acute pancreatitis, there is a correlation between
elevated serum PLA2 levels and inflammation and lipid metabolites are also
potential mediators in these events. In this regard it will be of interest to
see whether selective inhibition of PLA2 can be used as a therapeutic approach
against type 1 diabetes. Recently,
components of the sphingolipid pathways have been thought to be important
contributors to the pathophysiology of diabetes300-302. PLA2 hydrolysis of membrane phospholipids also generates a
lysophospholipid (lysoPL). Several types of lysoPL's exist in the islet
membrane: lysophosphatidylcholine (lysoPC), -ethanolamine (lysoPE), -serine
(lysoPS) and -inositol (lysoPI). LysoPL's and phosphatidic acid (PA) are shown
to promote insulin release when the cellular content of either lipid increases234. LysoPL's affect stimulus-secretion coupling at a number of points247, 248, 303,
304. They have been observed to inhibit directly the KATP channel activity
in the pancreas305. LysoPC and lysophosphatidylglycerol can also
effectively mobilize Ca2+ from intracellular stores304. LysoPAF has been demonstrated to circumvent the
inhibition of glucose-induced insulin release caused by phospholipase
inhibitors306.
Micro RNA and Diabetes
There may be as many as 1,000
different micro RNAs in the human genome, Micro RNAs, following production in
the nucleus by the enzyme Drosha are exported to the cytoplasm, and processed
by the enzye DICER. They then are incorporated into the RNA-induced
silencing complex150 and function to either silence by binding to
messenger RNA or by directing the cleavage of mRNA. It is estimated that there
are 68 miRNAs in beta cells. MiRNAs play essential roles in many biological processes in
mammals, including insulin secretion307, beta-cell development308-311, and adipocyte
differentiation312. The function of one islet specific miRNA has recently
been elucidated. MiRNA-375 is reported to inhibit insulin secretion by
inhibiting the production of the protein myotrophin and thereby decreasing
granule exocytosis312. In fact
more MiRNAs have been recently reported to be enriched in the islets (miR-127-3p, miR-184, miR-195
and miR-493) of which many of them are differentially expressed in healthy and
glucose intolerant subjects313. Further support also comes from MiRna
that have been identified to play a role in insulin biosynthesis and signaling314-319.
Concluding
remarks
Despite considerable progress in defining the molecular physiology of insulin
secretion there remain many unanswered questions. It is also important to
recognize that some current techniques may introduce complexity, such as the
recent report that the RIP-Cre transgene itself produces mice with impaired
glucose tolerance320. Nevertheless given our current knowledge it is clear that the
pancreatic beta cell is an excitable cell that is critically dependent upon
oxidative metabolism 321 and dependent on the influx of Ca2+ through gated channels and its
subsequent removal from the cell by intracellular sequestration or plasma
membrane pumping leaves it in a precarious position when faced with agents that
disrupt Ca2+ homeostasis in the cell. In a similar way, its susceptibility to
drugs such as the DNA alkylating agent streptozotocin or the free radical
generator alloxan may be the consequence of the mechanism of sugar transport,
which can carry these agents into the cell. Such phenomenon may explain in part
why so many of the agents that one associates with pancreatic beta cell damage
and death turn out to be specific secretagogues at least in the short-term. The
challenge is to discover which interactions are part of the normal
responsiveness of the cell to metabolizable substrates, peptides and
neurotransmitters and which are truly deleterious and thus a legitimate target
for pharmacological intervention. It is likely that the concepts that there are
"good" or a "bad" cytokines in the context of autoimmune
diabetes is no more valid than that of glucose being "good" or
"bad" in the context of glucotoxicity and type 2 diabetes. The
short-term effects of secretagogues on islet function have been extensively
documented but there is also the need to consider the broader perspective of
how the cell responds in the longer term to inflammatory stimuli or
physiological changes in energy homeostasis such as accompany pregnancy and
starvation (Fig. 9).
Figure 9. Yin and Yan
representation of the pancreatic beta cell in terms of induction of either cell
death or secretory response to secretagogues such as inflammatory cytokines and
PLA2.
Reference List
1.
Eisenbarth GS. Type 1 diabetes:
molecular, cellular and clinical immunology. Adv Exp Med Biol 2004;552.
2.
Wucherpfennig KW. Insights into
autoimmunity gained from structural analysis of MHC-peptide complexes. Curr
Opin Immunol 2001;13(6).
3.
Yoon JW, Jun HS. Cellular and
molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann N Y
Acad Sci 2001;928.
4.
Rosmalen JG, Leenen PJ, Pelegri C,
Drexhage HA, Homo-Delarche F. Islet abnormalities in the pathogenesis of
autoimmune diabetes. Trends Endocrinol Metab 2002;13(5).
5.
Dib SA, Gomes MB. Etiopathogenesis
of type 1 diabetes mellitus: prognostic factors for the evolution of residual
beta cell function. Diabetol Metab Syndr 2009;1(1).
6.
Zhang L, Gianani R, Nakayama M et
al. Type 1 diabetes: chronic progressive autoimmune disease. Novartis Found
Symp 2008;292.
7.
Rossini AA. Autoimmune diabetes and
the circle of tolerance. Diabetes 2004;53(2).
8.
Arif S, Tree TI, Astill TP et al.
Autoreactive T cell responses show proinflammatory polarization in diabetes but
a regulatory phenotype in health. J Clin Invest 2004;113(3).
9.
Bluestone JA, Herold K, Eisenbarth
G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature
2010;464(7293).
10.
Llaurado G, Gallart L, Tirado R et
al. Insulin resistance, low-grade inflammation and type 1 diabetes mellitus.
Acta Diabetol 2012.
11.
Nadeau KJ, Regensteiner JG, Bauer
TA et al. Insulin resistance in adolescents with type 1 diabetes and its
relationship to cardiovascular function. J Clin Endocrinol Metab 2010;95(2).
12.
Rodrigues TC, Biavatti K, Almeida
FK, Gross JL. Coronary artery calcification is associated with insulin
resistance index in patients with type 1 diabetes. Braz J Med Biol Res
2010;43(11).
13.
Wilkin TJ. Insulin resistance and
progression to type 1 diabetes in the European Nicotinamide Diabetes
Intervention Trial (ENDIT): response to Bingley et al. Diabetes Care
2008;31(4).
14.
Wilkin TJ. The accelerator
hypothesis: a review of the evidence for insulin resistance as the basis for
type I as well as type II diabetes. Int J Obes (Lond) 2009;33(7).
15.
Atkinson MA, Eisenbarth GS. Type 1
diabetes: new perspectives on disease pathogenesis and treatment. Lancet
2001;358(9277).
16.
Butler AE, Galasso R, Meier JJ,
Basu R, Rizza RA, Butler PC. Modestly increased beta cell apoptosis but no
increased beta cell replication in recent-onset type 1 diabetic patients who
died of diabetic ketoacidosis. Diabetologia 2007;50(11):2323-2331.
17.
Meier JJ, Lin JC, Butler AE,
Galasso R, Martinez DS, Butler PC. Direct evidence of attempted beta cell
regeneration in an 89-year-old patient with recent-onset type 1 diabetes.
Diabetologia 2006;49(8):1838-1844.
18.
Atkinson MA, Gianani R. The
pancreas in human type 1 diabetes: providing new answers to age-old questions.
Curr Opin Endocrinol Diabetes Obes 2009;16(4):279-285.
19.
Wang L, Lovejoy NF, Faustman DL.
Persistence of Prolonged C-peptide Production in Type 1 Diabetes as Measured
With an Ultrasensitive C-peptide Assay. Diabetes Care 2012;35(3).
20.
Gianani R, Campbell-Thompson M,
Sarkar SA et al. Dimorphic histopathology of long-standing childhood-onset
diabetes. Diabetologia 2010;53(4).
21.
Keenan HA, Sun JK, Levine J et al.
Residual insulin production and pancreatic ss-cell turnover after 50 years of
diabetes: Joslin Medalist Study. Diabetes 2010;59(11):2846-2853.
22.
Foulis AK, Stewart JA. The pancreas
in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content
of islets, insulitis and associated changes in the exocrine acinar tissue.
Diabetologia 1984;26(6):456-461.
23.
Eisenbarth GS. Banting Lecture
2009: An Unfinished Journey: Molecular Pathogenesis to Prevention of Type 1A
Diabetes. Diabetes 2010;59(4):759-774.
24.
Henderson JR. Why are the islet of
Langerhans? Lancet 1969;2(7618):469-470.
25.
Williams JA, Goldfine ID. The
insulin-pancreatic acinar axis. Diabetes 1985;34(10):980-986.
26.
Williams AJ, Chau W, Callaway MP,
Dayan CM. Magnetic resonance imaging: a reliable method for measuring
pancreatic volume in Type 1 diabetes. Diabet Med 2007;24(1):35-40.
27.
Gaglia JL, Guimaraes AR,
Harisinghani M et al. Noninvasive imaging of pancreatic islet inflammation in
type 1A diabetes patients. J Clin Invest 2011;121(1):442-445.
28.
Jiao Y, Peng ZH, Xing TH, Qin J,
Zhong CP. Assessment of islet graft survival using a 3.0-Tesla magnetic
resonance scanner. Anat Rec (Hoboken) 2008;291(12).
29.
Wang P, Yigit MV, Medarova Z et al.
Combined small interfering RNA therapy and in vivo magnetic resonance imaging
in islet transplantation. Diabetes 2011;60(2).
30.
Altieri DC. Molecular cloning of
effector cell protease receptor-1, a novel cell surface receptor for the
protease factor Xa. J Biol Chem 1994;269(5).
31.
Liggins C, Orlicky DJ, Bloomquist
LA, Gianani R. Developmentally regulated expression of Survivin in human
pancreatic islets. Pediatr Dev Pathol 2003;6(5).
32.
Hasel C, Bhanot UK, Heydrich R,
Strater J, Moller P. Parenchymal regression in chronic pancreatitis spares
islets reprogrammed for the expression of NFkappaB and IAPs. Lab Invest
2005;85(10).
33.
Achenbach P, Bonifacio E, Koczwara
K, Ziegler AG. Natural history of type 1 diabetes. Diabetes 2005;54 Suppl 2.
34.
Achenbach P, Bonifacio E, Ziegler
AG. Predicting type 1 diabetes. Curr Diab Rep 2005;5(2).
35.
Bingley PJ, Gale EA. Progression to
type 1 diabetes in islet cell antibody-positive relatives in the European Nicotinamide
Diabetes Intervention Trial: the role of additional immune, genetic and
metabolic markers of risk. diabetol 2006.
36.
Bottazzo GF, Bosi E, Bonifacio E,
Mirakian R, Todd I, Pujol-Borrell R. Pathogenesis of type I (insulin-dependent)
diabetes: possible mechanisms of autoimmune damage. Br Med Bull 1989;45(1).
37.
Eisenbarth GS. Italian Society of
Diabetology Mentor Award. The stages of Type 1A diabetes: retrospective and
prospective. Diabetes Nutr Metab 2004;17(6).
38.
Palmer JP, Fleming GA, Greenbaum CJ
et al. C-Peptide Is the Appropriate Outcome Measure for Type 1 Diabetes
Clinical Trials to Preserve beta-Cell Function: Report of an ADA Workshop,
21-22 October 2001. diab 2004;53(1).
39.
Andre I, Gonzalez A, Wang B, Katz
J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease:
lessons from diabetes models. Proc Natl Acad Sci U S A 1996;93(6).
40.
Bresson D, Togher L, Rodrigo E et
al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from
recent-onset autoimmune diabetes by inducing Tregs. Journal of Clinical
Investigation 2006;116(5).
41.
Dai YD, Jensen KP, Lehuen A et al.
A peptide of glutamic acid decarboxylase 65 can recruit and expand a
diabetogenic T cell clone, BDC2.5, in the pancreas. J Immunol 2005;175(6).
42.
Liu W, Putnam AL, Xu-Yu Z et al.
CD127 expression inversely correlates with FoxP3 and suppressive function of
human CD4+ T reg cells. J Exp Med 2006;203(7).
43.
Sarkar SA, Lee CE, Victorino F et
al. Expression and regulation of chemokines in murine and human type 1
diabetes. Diabetes 2012;61(2).
44.
Slack JM. Developmental biology of
the pancreas. Development 1995;121(6).
45.
Piper K, Brickwood S, Turnpenny LW
et al. Beta cell differentiation during early human pancreas development. J
Endocrinol 2004;181(1).
46.
Richardson MK, Hanken J, Gooneratne
ML et al. There is no highly conserved embryonic stage in the vertebrates:
implications for current theories of evolution and development. Anat Embryol
(Berl) 1997;196(2).
47.
Sarkar SA, Kobberup S, Wong R et
al. Global gene expression profiling and histochemical analysis of the
developing human fetal pancreas. Diabetologia 2008;51(2).
48.
Reichert M, Rustgi AK. Pancreatic
ductal cells in development, regeneration, and neoplasia. J Clin Invest
2011;121(12).
49.
Iburi T, Izumiyama H, Hirata Y.
[Endocrine glands of pancreas]. Nihon Rinsho69 Suppl 2.
50.
Youos JG. The role of alpha-,
delta- and F cells in insulin secretion and action. Diabetes Res Clin Pract93
Suppl 1.
51.
Prado CL, Pugh-Bernard AE, Elghazi
L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells
in two mouse models of pancreas development. Proc Natl Acad Sci U S A
2004;101(9).
52.
Sorenson RL, Garry DG, Brelje TC.
Structural and functional considerations of GABA in islets of Langerhans.
Beta-cells and nerves. Diabetes 1991;40(11).
53.
Wiest-Ladenburger U, Hartmann R,
Hartmann U, Berling K, Bohm BO, Richter W. Combined analysis and single-step
detection of GAD65 and IA2 autoantibodies in IDDM can replace the histochemical
islet cell antibody test. Diabetes 1997;46(4).
54.
Aanstoot HJ, Kang SM, Kim J et al.
Identification and characterization of glima 38, a glycosylated islet cell
membrane antigen, which together with GAD65 and IA2 marks the early phases of
autoimmune response in type 1 diabetes. J Clin Invest 1996;97(12).
55.
Hawkes CJ, Wasmeier C, Christie MR,
Hutton JC. Identification of the 37-kDa antigen in IDDM as a tyrosine
phosphatase-like protein (phogrin) related to IA-2. diab 1996;45(9).
56.
Bartholomeusz RK, Campbell IL,
Harrison LC. Pancreatic islet A2B5- and 3G5-reactive gangliosides are markers
of differentiation in rat insulinoma cells. Endocrinology 1989;124(6).
57.
Alejandro R, Shienvold FL, Hajek
SAV, Pierce M, Paul R, Mintz DH. A ganglioside antigen on the rat pancreatic B
cell surface identified by monoclonal antibody R2D6. J Clin Invest 1984;74.
58.
Anlauf M, Eissele R, Schafer MK et
al. Expression of the two isoforms of the vesicular monoamine transporter
(VMAT1 and VMAT2) in the endocrine pancreas and pancreatic endocrine tumors. J
Histochem Cytochem 2003;51(8).
59.
Harris PE, Ferrara C, Barba P,
Polito T, Freeby M, Maffei A. VMAT2 gene expression and function as it applies
to imaging beta-cell mass. J Mol Med (Berl) 2008;86(1).
60.
Eipper BA, Green CB, Mains RE.
Expression of prohormone processing enzymes in neuroendocrine and
non-neuroendocrine cells. J Natl Cancer Inst Monogr 1992;(13).
61.
Eipper BA, Milgram SL, Husten EJ,
Yun HY, Mains RE. Peptidylglycine alpha-amidating monooxygenase: a
multifunctional protein with catalytic, processing, and routing domains.
Protein Sci 1993;2(4).
62.
Eipper BA, Perkins SN, Husten EJ,
Johnson RC, Keutmann HT, Mains RE. Peptidyl-alpha-hydroxyglycine
alpha-amidating lyase. Purification, characterization, and expression. J Biol
Chem 1991;266(12).
63.
Schafer MK, Stoffers DA, Eipper BA,
Watson SJ. Expression of peptidylglycine alpha-amidating monooxygenase (EC
1.14.17.3) in the rat central nervous system. J Neurosci 1992;12(1).
64.
Martinez A, Montuenga LM, Springall
DR, Treston A, Cuttitta F, Polak JM. Immunocytochemical localization of
peptidylglycine alpha-amidating monooxygenase enzymes (PAM) in human endocrine
pancreas. J Histochem Cytochem 1993;41(3).
65.
Orskov C, Bersani M, Johnsen AH,
Hojrup P, Holst JJ. Complete sequences of glucagon-like peptide-1 from human
and pig small intestine. J Biol Chem 1989;264(22).
66.
Ashcroft FM, Rorsman P.
Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 1989;54(2).
67.
Colombo C, Porzio O, Liu M et al.
Seven mutations in the human insulin gene linked to permanent
neonatal/infancy-onset diabetes mellitus. J Clin Invest 2008;118(6).
68.
Hodish I, Liu M, Rajpal G et al.
Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol
Chem 2011;285(1).
69.
Hodish I, Absood A, Liu L et al. In
vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes
2011;60(8).
70.
Atkinson MA, Bluestone JA,
Eisenbarth GS et al. How does type 1 diabetes develop?: the notion of homicide
or beta-cell suicide revisited. Diabetes 2011;60(5).
71.
Grimaldi KA, Siddle K, Hutton JC.
Biosynthesis of insulin secretory granule membrane proteins. Control by
glucose. Biochem J 1987;245(2).
72.
Orci L, Ravazzola M, Storch MJ,
Anderson RG, Vassalli JD, Perrelet A. Proteolytic maturation of insulin is a
post-Golgi event which occurs in acidifying clathrin-coated secretory vesicles.
Cell 1987;49(6).
73.
Hutton JC, Davidson HW, Peshavaria
M. Proteolytic processing of chromogranin A in purified insulin granules.
Formation of a 20 kDa N-terminal fragment (betagranin) by the concerted action
of a Ca2+-dependent endopeptidase and carboxypeptidase H (EC 3.4.17.10).
Biochem J 1987;244(2).
74.
Orci L, Ravazzola M, Amherdt M et
al. Conversion of proinsulin to insulin occurs coordinately with acidification
of maturing secretory vesicles. J Cell Biol 1986;103(6 Pt 1).
75.
Orci L, Ravazzola M, Perrelet A.
(Pro)insulin associates with Golgi membranes of pancreatic B cells. Proc Natl
Acad Sci U S A 1984;81(21).
76.
Lazure C, Seidah NG, Pelaprat D,
Chretien M. Proteases and posttranslational processing of prohormones: a
review. Can J Biochem Cell Biol 1983;61(7).
77.
Lernmark A, Chan SJ, Choy R et al.
Biosynthesis of insulin and glucagon: a view of the current state of the art.
Ciba Found Symp 1976;41.
78.
Dodson G, Steiner D. The role of
assembly in insulin's biosynthesis. Curr Opin Struct Biol 1998;8(2).
79.
Emdin SO, Dodson GG, Cutfield JM,
Cutfield SM. Role of zinc in insulin biosynthesis. Some possible zinc-insulin
interactions in the pancreatic B-cell. Diabetologia 1980;19(3).
80.
Clifford KS, MacDonald MJ. Survey
of mRNAs encoding zinc transporters and other metal complexing proteins in
pancreatic islets of rats from birth to adulthood: similar patterns in the
Sprague-Dawley and Wistar BB strains. Diabetes Res Clin Pract 2000;49(2-3).
81.
Vallee BL, Falchuk KH. The
biochemical basis of zinc physiology. Physiol Rev 1993;73(1).
82.
Chimienti F, Aouffen M, Favier A,
Seve M. Zinc homeostasis-regulating proteins: new drug targets for triggering
cell fate. Curr Drug Targets 2003;4(4).
83.
Bloc A, Cens T, Cruz H, Dunant Y.
Zinc-induced changes in ionic currents of clonal rat pancreatic -cells:
activation of ATP-sensitive K+ channels. J Physiol 2000;529 Pt 3.
84.
Franklin I, Gromada J, Gjinovci A,
Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell
ATP-dependent potassium channels to inhibit glucagon release. Diabetes
2005;54(6).
85.
Ishihara H, Maechler P, Gjinovci A,
Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release
from neighbouring alpha-cells. Nat Cell Biol 2003;5(4).
86.
Kim BJ, Kim YH, Kim S et al. Zinc
as a paracrine effector in pancreatic islet cell death. Diabetes 2000;49(3).
87.
Kambe T, Narita H, Yamaguchi-Iwai Y
et al. Cloning and characterization of a novel mammalian zinc transporter, zinc
transporter 5, abundantly expressed in pancreatic beta cells. J Biol Chem
2002;277(21).
88.
Eide DJ. Metal ion transport in
eukaryotic microorganisms: insights from Saccharomyces cerevisiae. Adv Microb
Physiol 2000;43.
89.
Seve M, Chimienti F, Devergnas S,
Favier A. In silico identification and expression of SLC30 family genes: an
expressed sequence tag data mining strategy for the characterization of zinc
transporters' tissue expression. BMC Genomics 2004;5(1).
90.
Sim BK, Fogler WE, Zhou XH et al.
Zinc ligand-disrupted recombinant human Endostatin: potent inhibition of tumor
growth, safety and pharmacokinetic profile. Angiogenesis 1999;3(1).
91.
Sim DL, Yeo WM, Chow VT. The novel
human HUEL (C4orf1) protein shares homology with the DNA-binding domain of the
XPA DNA repair protein and displays nuclear translocation in a cell
cycle-dependent manner. Int J Biochem Cell Biol 2002;34(5).
92.
Palmiter RD, Findley SD. Cloning
and functional characterization of a mammalian zinc transporter that confers
resistance to zinc. EMBO J 1995;14(4).
93.
Wenzel HJ, Cole TB, Born DE,
Schwartzkroin PA, Palmiter RD. Ultrastructural localization of zinc
transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons
in the hippocampus of mouse and monkey. Proc Natl Acad Sci U S A 1997;94(23).
94.
Wenzlau JM, Juhl K, Yu L et al. The
cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1
diabetes. Proc Natl Acad Sci U S A 2007;104(43).
95.
Chimienti F, Devergnas S, Favier A,
Seve M. Identification and cloning of a beta-cell-specific zinc transporter,
ZnT-8, localized into insulin secretory granules. Diabetes 2004;53(9).
96.
Chimienti F, Devergnas S, Pattou F
et al. In vivo expression and functional characterization of the zinc
transporter ZnT8 in glucose-induced insulin secretion. J Cell Sci 2006;119(Pt
20).
97.
An S, Zenisek D. Regulation of
exocytosis in neurons and neuroendocrine cells. Curr Opin Neurobiol 2004;14(5).
98.
Tsuboi T, McMahon HT, Rutter GA.
Mechanisms of dense core vesicle recapture following "kiss and run"
("cavicapture") exocytosis in insulin-secreting cells. J Biol Chem
2004;279(45).
99.
Artalejo CR, Elhamdani A, Palfrey
HC. Secretion: dense-core vesicles can kiss-and-run too. Curr Biol 1998;8(2).
100.
Wu LG. Kinetic regulation of
vesicle endocytosis at synapses. Trends Neurosci 2004;27(9).
101.
Artalejo CR, Elhamdani A, Palfrey
HC. Sustained stimulation shifts the mechanism of endocytosis from
dynamin-1-dependent rapid endocytosis to clathrin- and dynamin-2-mediated slow
endocytosis in chromaffin cells. Proc Natl Acad Sci U S A 2002;99(9).
102.
Artalejo CR, Henley JR, McNiven MA,
Palfrey HC. Rapid endocytosis coupled to exocytosis in adrenal chromaffin cells
involves Ca2+, GTP, and dynamin but not clathrin. Proc Natl Acad Sci U S A
1995;92(18).
103.
Straub SG, Shanmugam G, Sharp GW.
Stimulation of insulin release by glucose is associated with an increase in the
number of docked granules in the beta-cells of rat pancreatic islets. Diabetes
2004;53(12).
104.
Straub SG, Sharp GW. Hypothesis:
one rate-limiting step controls the magnitude of both phases of
glucose-stimulated insulin secretion. Am J Physiol Cell Physiol 2004;287(3).
105.
Daniel S, Noda M, Straub SG, Sharp
GW. Identification of the docked granule pool responsible for the first phase
of glucose-stimulated insulin secretion. Diabetes 1999;48(9).
106.
Leibiger B, Leibiger IB, Moede T et
al. Selective insulin signaling through A and B insulin receptors regulates
transcription of insulin and glucokinase genes in pancreatic beta cells. Mol
Cell 2001;7(3).
107.
Leibiger IB, Leibiger B, Moede T,
Berggren PO. Exocytosis of insulin promotes insulin gene transcription via the
insulin receptor/PI-3 kinase/p70 s6 kinase and CaM kinase pathways. Mol Cell
1998;1(6).
108.
Schuit FC, In't Veld PA, Pipeleers
DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment
of pancreatic beta cells. Proc Natl Acad Sci U S A 1988;85(11).
109.
Fonseca SG, Fukuma M, Lipson KL et
al. WFS1 is a novel component of the unfolded protein response and maintains
homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem
2005;280(47).
110.
Hostens K, Pavlovic D, Zambre Y et
al. Exposure of human islets to cytokines can result in disproportionately
elevated proinsulin release. J Clin Invest 1999;104(1).
111.
Cnop M, Welsh N, Jonas JC, Jorns A,
Lenzen S, Eizirik DL. Mechanisms of Pancreatic {beta}-Cell Death in Type 1 and
Type 2 Diabetes: Many Differences, Few Similarities. Diabetes 2005;54 Suppl
2:S97-S107.
112.
Eizirik DL, Mandrup-Poulsen T. A
choice of death - the signal-transduction of immune-mediated beta-cell
apoptosis. diabetol 2001;44(12).
113.
Schuit FC, Huypens P, Heimberg H,
Pipeleers DG. Glucose sensing in pancreatic beta-cells: a model for the study
of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes
2001;50(1).
114.
Matschinsky FM. Banting Lecture
1995. A lesson in metabolic regulation inspired by the glucokinase glucose
sensor paradigm. Diabetes 1996;45(2).
115.
Schuit F, De Vos A, Farfari S et
al. Metabolic fate of glucose in purified islet cells. Glucose-regulated
anaplerosis in beta cells. J Biol Chem 1997;272(30).
116.
Drucker DJ. The biology of incretin
hormones. Cell Metab 2006;3(3).
117.
Hay CW, Sinclair EM, Bermano G,
Durward E, Tadayyon M, Docherty K. Glucagon-like peptide-1 stimulates human
insulin promoter activity in part through cAMP-responsive elements that lie
upstream and downstream of the transcription start site. J Endocrinol
2005;186(2).
118.
Park S, Dong X, Fisher TL et al.
Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and
function. J Biol Chem 2006;281(2).
119.
Triplitt C, Wright A, Chiquette E.
Incretin mimetics and dipeptidyl peptidase-IV inhibitors: potential new
therapies for type 2 diabetes mellitus. Pharmacotherapy 2006;26(3).
120.
de Sa SV, Correa-Giannella ML,
Machado MC et al. Somatostatin receptor subtype 5 (SSTR5) mRNA expression is
related to histopathological features of cell proliferation in insulinomas.
Endocr Relat Cancer 2006;13(1).
121.
Degano P, Peiro E, Miralles P,
Silvestre RA, Marco J. Effects of rat pancreatic polypeptide on islet-cell
secretion in the perfused rat pancreas. Metabolism 1992;41(3).
122.
Lundquist I, Sundler F, Ahren B,
Alumets J, Hakanson R. Somatostatin, pancreatic polypeptide, substance P, and
neurotensin: cellular distribution and effects on stimulated insulin secretion
in the mouse. Endocrinology 1979;104(3).
123.
Szecowka J, Tatemoto K, Rajamaki G,
Efendic S. Effects of PYY and PP on endocrine pancreas. Acta Physiol Scand
1983;119(2).
124.
Doi A, Shono T, Nishi M, Furuta H,
Sasaki H, Nanjo K. IA-2beta, but not IA-2, is induced by ghrelin and inhibits
glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A 2006;103(4).
125.
Takahashi H, Kurose Y, Kobayashi S
et al. Ghrelin enhances glucose-induced insulin secretion in scheduled meal-fed
sheep. J Endocrinol 2006;189(1).
126.
Henquin JC. Triggering and
amplifying pathways of regulation of insulin secretion by glucose. Diabetes
2000;49(11).
127.
Stiernet P, Guiot Y, Gilon P,
Henquin JC. Glucose acutely decreases pH of secretory granules in mouse
pancreatic islets: mechanisms and influence on insulin secretion. J Biol Chem
2006;.
128.
Johnson KH, O'Brien TD, Betsholtz
C, Westermark P. Islet amyloid polypeptide: mechanisms of amyloidogenesis in
the pancreatic islets and potential roles in diabetes mellitus. Lab Invest
1992;66.
129.
Juan-Pico P, Fuentes E, Javier
Bermudez-Silva F et al. Cannabinoid receptors regulate Ca(2+) signals and
insulin secretion in pancreatic beta-cell. Cell Calcium 2006;39(2).
130.
Kubosaki A, Nakamura S, Notkins AL.
Dense Core Vesicle Proteins IA-2 and IA-2{beta}: Metabolic Alterations in
Double Knockout Mice. diab 2005;54 Suppl 2.
131.
Moriyama H, Abiru N, Paronen J et
al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis
and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A
2003;100(18).
132.
Nakayama M, Abiru N, Moriyama H et
al. Prime role for an insulin epitope in the development of type 1 diabetes in
NOD mice. Nature 2005;435(7039).
133.
Han B, Serra P, Amrani A et al.
Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high
efficiency of a low-affinity peptide. Nat Med 2005;11(6).
134.
Rotig A, Cormier V, Chatelain P, et
al. Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus,
optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest
1993;91.
135.
Rabinovitch A, Pukel C, Baquerizo
H. Interleukin-1 inhibits glucose-modulated insulin and glucagon secretion in
rat islet monolayer cultures. Endocrinology 1988;122(6).
136.
Chen MC, Paez-Espinosa V, Welsh N,
Eizirik DL. Interleukin-1beta regulates phospholipase D-1 expression in rat
pancreatic beta-cells. Endocrinology 2000;141(8).
137.
Nielsen K, Karlsen AE, Deckert M et
al. Beta-cell maturation leads to in vitro sensitivity to cytotoxins. Diabetes
1999;48(12).
138.
Strandell E, Buschard K, Saldeen J, Welsh
N. Interleukin-1 beta induces the expression of hsp70, heme oxygenase and
Mn-SOD in FACS-purified rat islet beta-cells, but not in alpha-cells. Immunol
Lett 1995;48(2).
139.
Horio F, Fukuda M, Katoh H et al.
Reactive oxygen intermediates in autoimmune islet cell destruction of the NOD
mouse induced by peritoneal exudate cells (rich in macrophages) but not T
cells. Diabetologia 1994;37(1).
140.
Jorns A, Tiedge M, Lenzen S, Munday
R. Effect of superoxide dismutase, catalase, chelating agents, and free radical
scavengers on the toxicity of alloxan to isolated pancreatic islets in vitro.
Free Radic Biol Med 1999;26(9-10).
141.
Eizirik DL, Sandler S, Welsh N,
Juntti-Berggren L, Berggren PO. Interleukin-1 beta-induced stimulation of
insulin release in mouse pancreatic islets is related to diacylglycerol
production and protein kinase C activation. Mol Cell Endocrinol 1995;111(2).
142.
Karlsen AE, Sparre T, Nielsen K,
Nerup J, Pociot F. Proteome analysis--a novel approach to understand the
pathogenesis of Type 1 diabetes mellitus. Dis Markers 2001;17(4).
143.
Karlsen AE, Storling ZM, Sparre T
et al. Immune-mediated beta-cell destruction in vitro and in vivo-A pivotal
role for galectin-3. Biochem Biophys Res Commun 2006;344(1).
144.
Eizirik DL. Interleukin-1 induced
impairment in pancreatic islet oxidative metabolism of glucose is potentiated
by tumor necrosis factor. Acta Endocrinol (Copenh) 1988;119(3).
145.
Miura M, Endo S, Kaku LL et al.
Plasma type II phospholipase A2 levels in patients with acute pancreatitis. Res
Commun Mol Pathol Pharmacol 2001;109(3-4).
146.
Uhl W, Schrag HJ, Schmitter N,
Aufenanger J, Nevalainen TJ, Buchler MW. Experimental study of a novel
phospholipase A2 inhibitor in acute pancreatitis. Br J Surg 1998;85(5).
147.
Gyulkhandanyan AV, Lee SC,
Bikopoulos G, Dai F, Wheeler MB. The Zn2+-transporting Pathways in Pancreatic
beta-Cells: A ROLE FOR THE L-TYPE VOLTAGE-GATED Ca2+ CHANNEL. J Biol Chem
2006;281(14).
148.
Chimienti F, Favier A, Seve M.
ZnT-8, a pancreatic beta-cell-specific zinc transporter. Biometals 2005;18(4).
149.
Itoh Y, Hinuma S. GPR40, a free
fatty acid receptor on pancreatic beta cells, regulates insulin secretion.
Hepatol Res 2005.
150.
Srikanta S, Telen M, Posillico JT
et al. Monoclonal antibodies to a human islet cell surface glycoprotein: 4F2
and LC7-2. Endocrinology 1987;120(6).
151.
Kanai Y, Endou H. Heterodimeric
amino acid transporters: molecular biology and pathological and pharmacological
relevance. Curr Drug Metab 2001;2(4).
152.
Kim dK, Ahn SG, Park JC, Kanai Y,
Endou H, Yoon JH. Expression of L-type amino acid transporter 1 (LAT1) and 4F2
heavy chain (4F2hc) in oral squamous cell carcinoma and its precusor lesions.
Anticancer Res 2004;24(3a).
153.
Magnuson MA. Tissue-specific
regulation of glucokinase gene expression. J Cell Biochem 1992;48(2).
154.
Watada H, Kajimoto Y, Miyagawa J et
al. PDX-1 induces insulin and glucokinase gene expressions in alphaTC1 clone 6
cells in the presence of betacellulin. Diabetes 1996;45(12).
155.
Ferber S, BeltrandelRio H, Johnson
JH et al. GLUT-2 gene transfer into insulinoma cells confers both low and high
affinity glucose-stimulated insulin release. Relationship to glucokinase
activity. J Biol Chem 1994;269(15).
156.
German MS. Glucose sensing in
pancreatic islet beta cells: the key role of glucokinase and the glycolytic
intermediates. Proc Natl Acad Sci U S A 1993;90(5).
157.
Halban PA, Praz GA, Wollheim CB.
Abnormal glucose metabolism accompanies failure of glucose to stimulate insulin
release from a rat pancreatic cell line (RINm5F). Biochem J 1983;212(2).
158.
Khan A, Chandramouli V, Ostenson CG
et al. Glucose cycling is markedly enhanced in pancreatic islets of obese
hyperglycemic mice. Endocrinology 1990;126(5).
159.
Ling ZC, Khan A, Delauny F et al.
Increased glucocorticoid sensitivity in islet beta-cells: effects on glucose
6-phosphatase, glucose cycling and insulin release. Diabetologia 1998;41(6).
160.
Ebert DH, Bischof LJ, Streeper RS
et al. Structure and promoter activity of an islet-specific
glucose-6-phosphatase catalytic subunit-related gene. Diabetes 1999;48(3).
161.
Sekine N, Cirulli V, Regazzi R et
al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate
dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J
Biol Chem 1994;269(7).
162.
Giroix MH, Rasschaert J, Sener A et
al. Study of hexose transport, glycerol phosphate shuttle and Krebs cycle in
islets of adult rats injected with streptozotocin during the neonatal period.
Mol Cell Endocrinol 1992;83(2-3).
163.
Sener A, Malaisse WJ. Hexose
metabolism in pancreatic islets. Ca(2+)-dependent activation of the glycerol
phosphate shuttle by nutrient secretagogues. J Biol Chem 1992;267(19).
164.
Farfari S, Schulz V, Corkey B,
Prentki M. Glucose-regulated anaplerosis and cataplerosis in pancreatic
beta-cells: possible implication of a pyruvate/citrate shuttle in insulin
secretion. Diabetes 2000;49(5).
165.
Malaisse WJ, Hutton JC, Kawazu S,
Herchuelz A, Valverde I, Sener A. The stimulus-secretion coupling of
glucose-induced insulin release. XXXV. The links between metabolic and cationic
events. Diabetologia 1979;16(5).
166.
Newgard CB, McGarry JD. Metabolic
coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem
1995;64:689-719.
167.
DeFronzo RA, Prato SD. Insulin
resistance and diabetes mellitus. J Diabetes Complications 1996;10(5).
168.
Rorsman P, Bokvist K, Ammala C,
Eliasson L, Renstrom E, Gabel J. Ion channels, electrical activity and insulin
secretion. Diabete Metab 1994;20(2).
169.
Williams JA. Electrical correlates
of secretion in endocrine and exocrine cells. Fed Proc 1981;40(2).
170.
Ashcroft FM. K(ATP) channels and
insulin secretion: a key role in health and disease. Biochem Soc Trans
2006;34(Pt 2).
171.
Ashcroft FM, Rorsman P.
ATP-sensitive K+ channels: a link between B-cell metabolism and insulin
secretion. Biochem Soc Trans 1990;18(1).
172.
Ashcroft SJ, Ashcroft FM.
Properties and functions of ATP-sensitive K-channels. Cell Signal 1990;2(3).
173.
Bryan J, Aguilar-Bryan L. The ABCs
of ATP-sensitive potassium channels: more pieces of the puzzle. Curr Opin Cell
Biol 1997;9(4).
174.
Cook DL, Satin LS, Ashford ML,
Hales CN. ATP-sensitive K+ channels in pancreatic beta-cells. Spare-channel
hypothesis. Diabetes 1988;37(5).
175.
Loussouarn G, Pike LJ, Ashcroft FM,
Makhina EN, Nichols CG. Dynamic sensitivity of ATP-sensitive K(+) channels to
ATP. J Biol Chem 2001;276(31).
176.
Song DK, Ashcroft FM. ATP
modulation of ATP-sensitive potassium channel ATP sensitivity varies with the
type of SUR subunit. J Biol Chem 2001;276(10).
177.
Sakura H, Ashcroft FM.
Identification of four trp1 gene variants murine pancreatic beta-cells.
Diabetologia 1997;40(5).
178.
Gembal M, Detimary P, Gilon P, Gao
ZY, Henquin JC. Mechanisms by which glucose can control insulin release
independently from its action on adenosine triphosphate-sensitive K+ channels
in mouse B cells. J Clin Invest 1993;91(3).
179.
Gembal M, Gilon P, Henquin JC.
Evidence that glucose can control insulin release independently from its action
on ATP-sensitive K+ channels in mouse B cells. J Clin Invest 1992;89(4).
180.
Komatsu M, Noda M, Sharp GW.
Nutrient augmentation of Ca2+-dependent and Ca2+-independent pathways in
stimulus-coupling to insulin secretion can be distinguished by their guanosine
triphosphate requirements: studies on rat pancreatic islets. Endocrinology
1998;139(3).
181.
Komatsu M, Schermerhorn T, Noda M,
Straub SG, Aizawa T, Sharp GW. Augmentation of insulin release by glucose in
the absence of extracellular Ca2+: new insights into stimulus-secretion
coupling. Diabetes 1997;46(12).
182.
Komatsu M, Sharp GW, Aizawa T,
Hashizume K. Glucose stimulation of insulin release without an increase in
cytosolic free Ca2+ concentration: a possible involvement of GTP. Jpn J Physiol
1997;47 Suppl 1:S22-4.
183.
Li GD, Luo RH, Metz SA. Effects of
inhibitors of guanine nucleotide synthesis on membrane potential and cytosolic
free Ca2+ levels in insulin-secreting cells. Biochem Pharmacol 2000;59(5).
184.
Regazzi R, Li G, Ullrich S, Jaggi
C, Wollheim CB. Different requirements for protein kinase C activation and
Ca2+-independent insulin secretion in response to guanine nucleotides.
Endogenously generated diacylglycerol requires elevated Ca2+ for kinase C
insertion into membranes. J Biol Chem 1989;264(17).
185.
Ashcroft FM, Gribble FM. ATP-sensitive
K+ channels and insulin secretion: their role in health and disease.
Diabetologia 1999;42(8).
186.
Babenko AP, Aguilar-Bryan L, Bryan
J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol 1998;60:667-87.
187.
Polak M, Shield J. Neonatal
Diabetes Mellitus -- genetic aspects 2004. Pediatr Endocrinol Rev 2004;2(2).
188.
Miki T, Nagashima K, Seino S. The
structure and function of the ATP-sensitive K+ channel in insulin-secreting
pancreatic beta-cells. J Mol Endocrinol 1999;22(2).
189.
Suzuki M, Fujikura K, Inagaki N,
Seino S, Takata K. Localization of the ATP-sensitive K+ channel subunit Kir6.2
in mouse pancreas. Diabetes 1997;46(9).
190.
Bokvist K, Olsen HL, Hoy M et al.
Characterisation of sulphonylurea and ATP-regulated K+ channels in rat
pancreatic A-cells. Pflugers Arch 1999;438(4).
191.
Gopel SO, Kanno T, Barg S, Rorsman
P. Patch-clamp characterisation of somatostatin-secreting -cells in intact
mouse pancreatic islets. J Physiol 2000;528(Pt 3).
192.
Gopel SO, Kanno T, Barg S, Weng XG,
Gromada J, Rorsman P. Regulation of glucagon release in mouse -cells by KATP
channels and inactivation of TTX-sensitive Na+ channels. J Physiol 2000;528(Pt
3).
193.
Hattersley AT, Ashcroft FM.
Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes,
new scientific insights, and new therapy. diab 2005;54(9).
194.
Koster JC, Remedi MS, Dao C,
Nichols CG. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+
channels in neonatal diabetes: implications for pharmacogenomic therapy. diab
2005;54(9).
195.
Proks P, Girard C, Haider S et al.
A gating mutation at the internal mouth of the Kir6.2 pore is associated with
DEND syndrome. EMBO Rep 2005;6(5).
196.
Nestorowicz A, Glaser B, Wilson BA
et al. Genetic heterogeneity in familial hyperinsulinism [published erratum
appears in Hum Mol Genet 1998 Sep;7(9):1527]. Hum Mol Genet 1998;7(7).
197.
Flanagan S, Damhuis A, Banerjee I
et al. Partial ABCC8 gene deletion mutations causing diazoxide-unresponsive
hyperinsulinaemic hypoglycaemia. Pediatr Diabetes 2011.
198.
Flanagan SE, Kapoor RR, Banerjee I
et al. Dominantly acting ABCC8 mutations in patients with medically
unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet 2011;79(6).
199.
Ismail D, Smith VV, de Lonlay P et
al. Familial focal congenital hyperinsulinism. J Clin Endocrinol Metab
2011;96(1).
200.
Kapoor RR, Flanagan SE, James CT et
al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant
ABCC8/KCNJ11 mutations. Diabetologia 2011;54(10).
201.
Kumaran A, Kapoor RR, Flanagan SE,
Ellard S, Hussain K. Congenital hyperinsulinism due to a compound heterozygous
ABCC8 mutation with spontaneous resolution at eight weeks. Horm Res Paediatr
2010;73(4).
202.
James C, Kapoor RR, Ismail D,
Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet
2009;46(5).
203.
Kapoor RR, Flanagan SE, James C,
Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child
2009;94(6).
204.
Kapoor RR, James C, Hussain K.
Hyperinsulinism in developmental syndromes. Endocr Dev 2009;14.
205.
Poon M, Hussain K. Postprandial
hyperinsulinaemic hypoglycaemia and type 1 diabetes mellitus. BMJ Case Rep
2009;2009.
206.
Kapoor RR, James C, Flanagan SE,
Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency
and hyperinsulinemic hypoglycemia: characterization of a novel mutation and
severe dietary protein sensitivity. J Clin Endocrinol Metab 2009;94(7).
207.
Cuesta-Munoz AL, Huopio H,
Otonkoski T et al. Severe persistent hyperinsulinemic hypoglycemia due to a de
novo glucokinase mutation. Diabetes 2004;53(8).
208.
Osbak KK, Colclough K, Saint-Martin
C et al. Update on mutations in glucokinase (GCK), which cause maturity-onset
diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic
hypoglycemia. Hum Mutat 2009;30(11).
209.
Kawajiri M, Okano Y, Kuno M et al.
Unregulated insulin secretion by pancreatic beta cells in
hyperinsulinism/hyperammonemia syndrome: role of glutamate dehydrogenase,
ATP-sensitive potassium channel, and nonselective cation channel. Pediatr Res
2006;59(3).
210.
Chan CB, Saleh MC, Koshkin V,
Wheeler MB. Uncoupling protein 2 and islet function. diab 2004;53 Suppl 1.
211.
Joseph JW, Koshkin V, Saleh MC et
al. Free fatty acid-induced beta-cell defects are dependent on uncoupling
protein 2 expression. J Biol Chem 2004;279(49).
212.
Ann K, Kowalchyk JA, Loyet KM,
Martin TF. Novel Ca2+-binding protein (CAPS) related to UNC-31 required for
Ca2+-activated exocytosis. J Biol Chem 1997;272(32).
213.
Bokvist K, Eliasson L, Ammala C,
Renstrom E, Rorsman P. Co-localization of L-type Ca2+ channels and
insulin-containing secretory granules and its significance for the initiation
of exocytosis in mouse pancreatic B-cells. EMBO J 1995;14(1).
214.
Wiser O, Trus M, Hernandez A et al.
The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the
exocytotic machinery. Proc Natl Acad Sci U S A 1999;96(1).
215.
Nilius B, Hess P, Lansman JB, Tsien
RW. A novel type of cardiac calcium channel in ventricular cells. Nature
1985;316(6027).
216.
Gutnick MJ, Lux HD, Swandulla D,
Zucker H. Voltage-dependent and calcium-dependent inactivation of calcium
channel current in identified snail neurones. J Physiol 1989;412:197-220.
217.
Ammala C, Eliasson L, Bokvist K et
al. Activation of protein kinases and inhibition of protein phosphatases play a
central role in the regulation of exocytosis in mouse pancreatic beta cells.
Proc Natl Acad Sci U S A 1994;91(10).
218.
Dean PM. Ultrastructural
morphometry of the pancreatic -cell. Diabetologia 1973;9(2).
219.
Eliasson L, Renstrom E, Ding WG,
Proks P, Rorsman P. Rapid ATP-dependent priming of secretory granules precedes
Ca(2+)-induced exocytosis in mouse pancreatic B-cells. J Physiol 1997;503(Pt
2).
220.
Gromada J, Hoy M, Renstrom E et al.
CaM kinase II-dependent mobilization of secretory granules underlies
acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J
Physiol 1999;518(Pt 3).
221.
Rorsman P, Eliasson L, Renstrom E,
Gromada J, Barg S, Gopel S. The Cell Physiology of Biphasic Insulin Secretion.
News Physiol Sci 2000;15:72-77.
222.
Rorsman P. The pancreatic beta-cell
as a fuel sensor: an electrophysiologist's viewpoint. Diabetologia 1997;40(5).
223.
Huang JD, Brady ST, Richards BW et
al. Direct interaction of microtubule- and actin-based transport motors. Nature
1999;397(6716).
224.
Easom RA. Beta-granule transport
and exocytosis. Semin Cell Dev Biol 2000;11(4).
225.
Sollner T, Bennett MK, Whiteheart
SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro
that may correspond to sequential steps of synaptic vesicle docking,
activation, and fusion. Cell 1993;75(3).
226.
Rupnik M, Kreft M, Sikdar SK et al.
Rapid regulated dense-core vesicle exocytosis requires the CAPS protein. Proc
Natl Acad Sci U S A 2000;97(10).
227.
Fukui K, Yang Q, Cao Y et al. The
HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation.
Cell Metab 2005;2(6).
228.
Evans GJ, Barclay JW, Prescott GR
et al. Protein kinase B/Akt is a novel cysteine string protein kinase that
regulates exocytosis release kinetics and quantal size. J Biol Chem
2006;281(3).
229.
Jahn R, Sudhof TC. Membrane fusion
and exocytosis. Annu Rev Biochem 1999;68:863-911.
230.
Izumi T, Gomi H, Torii S.
Functional analysis of Rab27a effector granuphilin in insulin exocytosis.
Methods Enzymol 2005;403.
231.
Gomi H, Mizutani S, Kasai K,
Itohara S, Izumi T. Granuphilin molecularly docks insulin granules to the
fusion machinery. J Cell Biol 2005;171(1).
232.
Jacobsson G, Bean AJ, Scheller RH
et al. Identification of synaptic proteins and their isoform mRNAs in
compartments of pancreatic endocrine cells. Proc Natl Acad Sci U S A
1994;91(26).
233.
Lang J. Molecular mechanisms and
regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J
Biochem 1999;259(1-2).
234.
Lao G, Scheuss V, Gerwin CM et al.
Syntaphilin: a syntaxin-1 clamp that controls SNARE assembly. Neuron
2000;25(1).
235.
Fisher RJ, Pevsner J, Burgoyne RD.
Control of fusion pore dynamics during exocytosis by Munc18. Science
2001;291(5505).
236.
Huang X, Kang YH, Pasyk EA et al.
Ca(2+) influx and cAMP elevation overcame botulinum toxin A but not tetanus
toxin inhibition of insulin exocytosis. Am J Physiol Cell Physiol 2001;281(3).
237.
Land J, Zhang H, Vaidyanathan VV,
Sadoul K, Niemann H, Wollheim CB. Transient expression of botulinum neurotoxin
C1 light chain differentially inhibits calcium and glucose induced insulin
secretion in clonal beta-cells. FEBS Lett 1997;419(1).
238.
Li C, Ullrich B, Zhang JZ, Anderson
RG, Brose N, Sudhof TC. Ca(2+)-dependent and -independent activities of neural
and non-neural synaptotagmins. Nature 1995;375(6532).
239.
Loyet KM, Kowalchyk JA, Chaudhary
A, Chen J, Prestwich GD, Martin TF. Specific binding of phosphatidylinositol
4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a
potential phosphoinositide effector protein for regulated exocytosis. J Biol
Chem 1998;273(14).
240.
Broer A, Friedrich B, Wagner CA et
al. Association of 4F2hc with light chains LAT1, LAT2 or y+LAT2 requires
different domains. Biochem J 2001;355(Pt 3).
241.
Xu T, Ashery U, Burgoyne RD, Neher
E. Early requirement for alpha-SNAP and NSF in the secretory cascade in chromaffin
cells. EMBO J 1999;18(12).
242.
Clark JD, Lin LL, Kriz RW et al. A
novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent
translocation domain with homology to PKC and GAP. Cell 1991;65(6).
243.
Sharp JD, White DL, Chiou XG et al.
Molecular cloning and expression of human Ca(2+)-sensitive cytosolic
phospholipase A2. J Biol Chem 1991;266(23).
244.
Iezzi M, Escher G, Meda P et al.
Subcellular distribution and function of Rab3A, B, C, and D isoforms in
insulin-secreting cells. Mol Endocrinol 1999;13(2).
245.
Kajio H, Olszewski S, Rosner PJ,
Donelan MJ, Geoghegan KF, Rhodes CJ. A low-affinity Ca2+-dependent association
of calmodulin with the Rab3A effector domain inversely correlates with insulin
exocytosis. Diabetes 2001;50(9).
246.
Balsinde J, Balboa MA, Insel PA,
Dennis EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol
Toxicol 1999;39:175-89.
247.
Bauerfeind R, Takei K, De Camilli
P. Amphiphysin I is associated with coated endocytic intermediates and undergoes
stimulation-dependent dephosphorylation in nerve terminals. J Biol Chem
1997;272(49).
248.
Lai MM, Hong JJ, Ruggiero AM et al.
The calcineurin-dynamin 1 complex as a calcium sensor for synaptic vesicle
endocytosis. J Biol Chem 1999;274(37).
249.
Kang G, Chepurny OG, Malester B et
al. cAMP Sensor Epac As A Determinant Of ATP-Sensitive Potassium Channel
Activity In Human Pancreatic Beta Cells And Rat INS-1 Cells. J Physiol 2006.
250.
Waselle L, Gerona RR, Vitale N,
Martin TF, Bader MF, Regazzi R. Role of phosphoinositide signaling in the
control of insulin exocytosis. Mol Endocrinol 2005;19(12).
251.
Ammala C, Ashcroft FM, Rorsman P.
Calcium-independent potentiation of insulin release by cyclic AMP in single
beta-cells. Nature 1993;363(6427).
252.
Hamilton PB, Stevens JR, Gidley J,
Holz P, Gibson WC. A new lineage of trypanosomes from Australian vertebrates
and terrestrial bloodsucking leeches (Haemadipsidae). Int J Parasitol
2005;35(4).
253.
Renstrom E, Eliasson L, Rorsman P.
Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP
in mouse pancreatic B-cells. J Physiol 1997;502(Pt 1).
254.
Ashcroft FM, Proks P, Smith PA,
Ammala C, Bokvist K, Rorsman P. Stimulus-secretion coupling in pancreatic beta
cells. J Cell Biochem 1994;55 Suppl:54-65.
255.
Murakami K, Chan SY, Routtenberg A.
Protein kinase C activation by cis-fatty acid in the absence of Ca2+ and
phospholipids. J Biol Chem 1986;261(33).
256.
Tischfield JA. A reassessment of
the low molecular weight phospholipase A2 gene family in mammals. J Biol Chem
1997;272(28).
257.
Leslie CC. Properties and
regulation of cytosolic phospholipase A2. J Biol Chem 1997;272(27).
258.
Sapirstein A, Bonventre JV.
Specific physiological roles of cytosolic phospholipase A(2) as defined by gene
knockouts. Biochim Biophys Acta 2000;1488(1-2).
259.
Bonventre JV, Huang Z, Taheri MR et
al. Reduced fertility and postischaemic brain injury in mice deficient in
cytosolic phospholipase A2. Nature 1997;390(6660).
260.
Murakami M, Nakatani Y, Kuwata H,
Kudo I. Cellular components that functionally interact with signaling
phospholipase A(2)s. Biochim Biophys Acta 2000;1488(1-2).
261.
Balsinde J, Dennis EA. Function and
inhibition of intracellular calcium-independent phospholipase A2. J Biol Chem
1997;272(26).
262.
Winstead MV, Balsinde J, Dennis EA.
Calcium-independent phospholipase A(2): structure and function. Biochim Biophys
Acta 2000;1488(1-2).
263.
Ramanadham S, Ma Z, Arita H, Zhang
S, Turk J. Type IB secretory phospholipase A2 is contained in insulin secretory
granules of pancreatic islet beta-cells and is co-secreted with insulin from
glucose-stimulated islets. Biochim Biophys Acta 1998;1390(3).
264.
Dennis EA. The growing
phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem Sci
1997;22(1).
265.
Stafforini DM, McIntyre TM,
Zimmerman GA, Prescott SM. Platelet-activating factor acetylhydrolases. J Biol
Chem 1997;272(29).
266.
Loweth AC, Scarpello JH, Morgan NG.
Phospholipase A2 expression in human and rodent insulin-secreting cells. Mol
Cell Endocrinol 1995;112(2).
267.
Metz S, Holmes D, Robertson RP,
Leitner W, Draznin B. Gene expression of type I phospholipase A2 in pancreatic
beta cells. Regulation of mRNA levels by starvation or glucose excess. FEBS
Lett 1991;295(1-3).
268.
Niwa T, Matsukawa Y, Senda T,
Nimura Y, Hidaka H, Niki I. Acetylcholine activates intracellular movement of
insulin granules in pancreatic beta-cells via inositol trisphosphate-dependent
[correction of triphosphate-dependent] mobilization of intracellular Ca2+.
Diabetes 1998;47(11).
269.
Parker KJ, Jones PM, Hunton CH,
Persaud SJ, Taylor CG, Howell SL. Identification and localisation of a type IV
cytosolic phospholipase A2 in rat pancreatic beta-cells. J Mol Endocrinol 1996;17(1).
270.
Eddlestone GT. ATP-sensitive K
channel modulation by products of PLA2 action in the insulin-secreting HIT cell
line. Am J Physiol 1995;268(1 Pt 1).
271.
Gross RW, Ramanadham S, Kruszka KK,
Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion
independent phospholipase A2 activity selective for hydrolysis of arachidonate
which is stimulated by adenosine triphosphate and is specifically localized to
islet beta-cells. Biochemistry 1993;32(1).
272.
Kashiwagi M, Friess H, Uhl W et al.
Phospholipase A2 isoforms are altered in chronic pancreatitis. Ann Surg
1998;227(2).
273.
Seeds MC, Jones DF, Chilton FH,
Bass DA. Secretory and cytosolic phospholipases A2 are activated during TNF
priming of human neutrophils. Biochim Biophys Acta 1998;1389(3).
274.
Ma Z, Ramanadham S, Kempe K, Chi
XS, Ladenson J, Turk J. Pancreatic islets express a Ca2+-independent
phospholipase A2 enzyme that contains a repeated structural motif homologous to
the integral membrane protein binding domain of ankyrin. J Biol Chem
1997;272(17).
275.
Turk J, Wolf BA, Lefkowith JB,
Stump WT, McDaniel ML. Glucose-induced phospholipid hydrolysis in isolated
pancreatic islets: quantitative effects on the phospholipid content of
arachidonate and other fatty acids. Biochim Biophys Acta 1986;879(3).
276.
Ma Z, Zhang S, Turk J, Ramanadham
S. Stimulation of insulin secretion and associated nuclear accumulation of
iPLA(2)beta in INS-1 insulinoma cells. Am J Physiol Endocrinol Metab
2002;282(4).
277.
Wolf BA, Pasquale SM, Turk J. Free
fatty acid accumulation in secretagogue-stimulated pancreatic islets and
effects of arachidonate on depolarization-induced insulin secretion.
Biochemistry 1991;30(26).
278.
Wolf BA, Turk J, Sherman WR,
McDaniel ML. Intracellular Ca2+ mobilization by arachidonic acid. Comparison
with myo-inositol 1,4,5-trisphosphate in isolated pancreatic islets. J Biol
Chem 1986;261(8).
279.
Bell RL, Stanford N, Kennerly DA,
Majerus PW. Diglyceride lipase: a pathway for arachidonate release from human
platelets. Adv Prostaglandin Thromboxane Res 1980;6:219-24.
280.
Konrad RJ, Major CD, Wolf BA.
Diacylglycerol hydrolysis to arachidonic acid is necessary for insulin
secretion from isolated pancreatic islets: sequential actions of diacylglycerol
and monoacylglycerol lipases. Biochemistry 1994;33(45).
281.
Konrad RJ, Jolly YC, Major C, Wolf
BA. Inhibition of phospholipase A2 and insulin secretion in pancreatic islets.
Biochim Biophys Acta 1992;1135(2).
282.
Metz SA. Pancreatic islet
phospholipase A2: differential, Ca2+-dependent effects of lysophospholipids,
arachidonic acid, and its lipoxygenase-derived metabolites on insulin release.
Adv Prostaglandin Thromboxane Leukot Res 1987;17B:668-76.
283.
Damron DS, Van Wagoner DR, Moravec
CS, Bond M. Arachidonic acid and endothelin potentiate Ca2+ transients in rat
cardiac myocytes via inhibition of distinct K+ channels. J Biol Chem
1993;268(36).
284.
Graber MN, Alfonso A, Gill DL. Ca2+
pools and cell growth: arachidonic acid induces recovery of cells growth-arrested
by Ca2+ pool depletion. J Biol Chem 1996;271(2).
285.
McPhail LC, Clayton CC, Snyderman
R. A potential second messenger role for unsaturated fatty acids: activation of
Ca2+-dependent protein kinase. Science 1984;224(4649).
286.
Shuttleworth TJ. Arachidonic acid
activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J Biol
Chem 1996;271(36).
287.
Metz SA. Exogenous arachidonic acid
promotes insulin release from intact or permeabilized rat islets by dual
mechanisms. Putative activation of Ca2+ mobilization and protein kinase C.
Diabetes 1988;37(11).
288.
Metz SA. The pancreatic islet as
Rubik's Cube. Is phospholipid hydrolysis a piece of the puzzle? Diabetes
1991;40(12).
289.
Ramanadham S, Gross R, Turk J.
Arachidonic acid induces an increase in the cytosolic calcium concentration in
single pancreatic islet beta cells. Biochem Biophys Res Commun 1992;184(2).
290.
Hirabayashi T, Shimizu T.
Localization and regulation of cytosolic phospholipase A(2). Biochim Biophys
Acta 2000;1488(1-2).
291.
Kimple ME, Nixon AB, Kelly P et al.
A role for G(z) in pancreatic islet beta-cell biology. J Biol Chem
2005;280(36).
292.
Kato R, Yamamoto S, Nakadate T,
Nakaki T. Possible involvement of phospholipase A2 activation and lipoxygenase
product(s) in the mechanism of insulin secretion. Adv Prostaglandin Thromboxane
Leukot Res 1983;12:265-70.
293.
Zhou YP, Teng D, Dralyuk F et al.
Apoptosis in insulin-secreting cells. Evidence for the role of intracellular
Ca2+ stores and arachidonic acid metabolism. J Clin Invest 1998;101(8).
294.
Frossard JL, Bhagat L, Lee HS et
al. Both thermal and non-thermal stress protect against caerulein induced
pancreatitis and prevent trypsinogen activation in the pancreas. Gut
2002;50(1).
295.
Ramsingh AI, Chapman N, Tracy S.
Coxsackieviruses and diabetes. Bioessays 1997;19(9).
296.
Sevilla N, Homann D, von Herrath M
et al. Virus-induced diabetes in a transgenic model: role of cross-reacting
viruses and quantitation of effector T cells needed to cause disease. J Virol
2000;74(7).
297.
Gu D, Sarvetnick N. Epithelial cell
proliferation and islet neogenesis in IFN-g transgenic mice. Development
1993;118(1).
298.
Pruzanski W, Vadas P. Phospholipase
A2--a mediator between proximal and distal effectors of inflammation. Immunol
Today 1991;12(5).
299.
van Puijenbroek AA, Wissink S, van
der Saag PT, Peppelenbosch MP. Phospholipase A2 inhibitors and leukotriene
synthesis inhibitors block TNF-induced NF-kappaB activation. Cytokine
1999;11(2).
300.
Deevska GM, Nikolova-Karakashian
MN. The twists and turns of sphingolipid pathway in glucose regulation.
Biochimie 2011;93(1).
301.
Fox TE, Bewley MC, Unrath KA et al.
Circulating sphingolipid biomarkers in models of type 1 diabetes. J Lipid Res
2011;52(3).
302.
Ortsater H. Arachidonic acid fights
palmitate: new insights into fatty acid toxicity in beta-cells. Clin Sci (Lond)
2011;120(5).
303.
Metz SA. Mobilization of cellular
Ca2+ by lysophospholipids in rat islets of Langerhans. Biochim Biophys Acta
1988;968(2).
304.
Rustenbeck I, Lenzen S. Effects of
lysophosphatidylcholine and arachidonic acid on the regulation of intracellular
Ca2+ transport. Naunyn Schmiedebergs Arch Pharmacol 1989;339(1-2).
305.
Gasser KW, Holda JR. ATP-sensitive
potassium transport by pancreatic secretory granule membrane. Am J Physiol
1993;264(1 Pt 1).
306.
Metz SA. Ether-linked
lysophospholipids initiate insulin secretion. Lysophospholipids may mediate
effects of phospholipase A2 activation on hormone release. Diabetes 1986;35(7).
307.
Fernandez-Valverde SL, Taft RJ,
Mattick JS. MicroRNAs in beta-cell biology, insulin resistance, diabetes and
its complications. Diabetes 2012;60(7).
308.
Joglekar MV, Parekh VS, Hardikar
AA. New pancreas from old: microregulators of pancreas regeneration. Trends
Endocrinol Metab 2007;18(10).
309.
Joglekar MV, Parekh VS, Mehta S,
Bhonde RR, Hardikar AA. MicroRNA profiling of developing and regenerating
pancreas reveal post-transcriptional regulation of neurogenin3. Dev Biol
2007;311(2).
310.
Joglekar MV, Joglekar VM, Hardikar
AA. Expression of islet-specific microRNAs during human pancreatic development.
Gene Expr Patterns 2009;9(2).
311.
Joglekar MV, Parekh VS, Hardikar
AA. Islet-specific microRNAs in pancreas development, regeneration and diabetes.
Indian J Exp Biol 2011;49(6).
312.
Cuellar TL, McManus MT. MicroRNAs
and endocrine biology. J Endocrinol 2005;187(3).
313.
Bolmeson C, Esguerra JL, Salehi A,
Speidel D, Eliasson L, Cilio CM. Differences in islet-enriched miRNAs in
healthy and glucose intolerant human subjects. Biochem Biophys Res
Commun404(1).
314.
Davalos A, Goedeke L, Smibert P et
al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin
signaling. Proc Natl Acad Sci U S A108(22).
315.
Fred RG, Bang-Berthelsen CH,
Mandrup-Poulsen T, Grunnet LG, Welsh N. High glucose suppresses human islet
insulin biosynthesis by inducing miR-133a leading to decreased polypyrimidine
tract binding protein-expression. PLoS One5(5).
316.
Frost RJ, Olson EN. Control of glucose
homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl
Acad Sci U S A108(52).
317.
Gallagher IJ, Scheele C, Keller P
et al. Integration of microRNA changes in vivo identifies novel molecular
features of muscle insulin resistance in type 2 diabetes. Genome Med2(2).
318.
Hennessy E, Clynes M, Jeppesen PB,
O'Driscoll L. Identification of microRNAs with a role in glucose stimulated
insulin secretion by expression profiling of MIN6 cells. Biochem Biophys Res
Commun396(2).
319.
Kalis M, Bolmeson C, Esguerra JL et
al. Beta-cell specific deletion of Dicer1 leads to defective insulin secretion
and diabetes mellitus. PLoS One6(12).
320.
Lee JY, Ristow M, Lin X, White MF,
Magnuson MA, Hennighausen L. RIP-Cre revisited, evidence for impairments of
pancreatic beta-cell function. J Biol Chem 2006;281(5).
321.
Wiederkehr A, Wollheim CB.
Implication of mitochondria in insulin secretion and action. Endocrinology
2006.