Chapter 3
(4/29/2011)
Animal Models of Type 1 Diabetes:
Genetics
and Immunological Function
George S. Eisenbarth
Modified from second web edition Lang J and Bellgrau
D
Introduction
The
past two decades has seen remarkable advances in understanding the genetics and
pathophysiology of spontaneous animal models of immune mediated diabetes (Type
1A) including structural characterization of class II MHC presentation of insulin
peptide B:9-231 as well as the chromagranin peptide WE-14 (previously elusive target of
the BDC2.5 T cell receptor2), the creation of new animal models 3 with either genetic manipulation of autoantigens4, 5 or immunologic function (e.g.,
toll receptor activation; peptide immunization, mutations of regulatory
pathways such as AIRE6 or foxP37), or a combination of genetic and environmental manipulation 8-18. “Humanized” mouse strains are being created and studied and promise
additional insights relevant to type 1A diabetes 19-21. In addition at the
Spontaneous
type 1 diabetes-susceptible models include the non-obese diabetic (NOD) mouse,
the BioBreeding Diabetes-Prone (BB-DP) rat, the Komeda Diabetes-Prone (KDP)
sub-line of the Long-Evans Tokushima Lean rat Lew.1.WR1 and the Lew.1AR1/Ztm
rat. Multiple experimentally-induced models of type 1 diabetes are available
including: 1) T cell receptor (TCR) transgenic (Tg) and retrogenic mice with
the T cell receptors of naturally occurring diabetogenic clones 2) Neo-antigen
(Ag) expression under the control of the rat insulin promoter (RIP) to
establish neo-self antigen pancreatic expression that can be the target of
autoimmunity, and 3) RIP-driven expression of costimulatory molecules on beta
cells. Mice with knockouts of putative
islet autoantigens have allowed direct testing of the pathogenic significance
of specific target molecules. Strains of mice with mutations of genes
associated with type 1 diabetes in man (FoxP3 and AIRE) are being studied (including
an autosomal dominant “human” AIRE mutation6). Such strains usually do not
develop diabetes, but rather have novel autoimmune phenotypes 22-24.
A major conclusion
from these models is that type 1 diabetes is not the result of a single pathway
but can be the result of numerous distinct mechanisms 25. Nonetheless, some generalities do exist including the polygenic nature
of the disease, the involvement of T cells in the destruction of pancreatic
beta cells, and the incomplete penetrance of the disease implying that
environmental or “stochastic” factors influence disease susceptibility. These
models provide useful tools for studying the autoimmune process as well as testing
potential treatments for the prevention of type 1 diabetes. There has been some
pessimism26, appropriately expressed 10, that therapies effective in animal models such as the NOD mouse may
not be directly relevant to man. No doubt the usual animal models are inbred
(thus not diallelic at any locus) and finely “balanced” in terms of progression
to diabetes while raised in pathogen-free environments. Thus, it is likely that
many more therapies will be effective in the animal models compared to man (Entelos
has developed a computer model of NOD mice and catalogued therapeutics27). Nevertheless, the initial
experimental success of anti-CD3 monoclonal antibody therapy in man to delay
loss of insulin secretion is a direct result of studies in the NOD mouse 28, and large trials such as the Diabetes Prevention Trial (DPT) have
provided preliminary data that oral insulin (also first studied in the NOD
mouse) in subjects with elevated insulin autoantibodies may delay disease
progression to diabetes29. We suspect that informed optimism relative to the utility of animal
models is in order26 , and that the more robust the therapy in the animal model (e.g.,
ability to abrogate insulitis, ability to prevent diabetes in models engineered
to be more highly penetrant [e.g., insulin 2 gene knockout NOD mice 30) , reversal of hyperglycemia, and the closer the human studies mimic
the animal studies (e.g., dose and timing), the more relevant the study will be
to human type 1 diabetes.
The Nonobese Diabetic Mouse
There are now more than 6,000 articles listed in PubMed
on the NOD mouse making any comprehensive review of the model a difficult task
with the certainty of failure to cite all important contributions and a
certainty that not all the different pathways being pursued will be
covered. Given the ability to
electronically access primary articles through the internet with services such
as PubMed and Google my purpose in this review on such a large topic is to
provide an overall organization, a particular viewpoint (with caveats), and to attempt
to highlight many important “relevant” observations. The general hypothesis my laboratory group is
pursuing is that NOD mice 1. have relatively mild defects of immune tolerance
(particularly in comparison to foxP3 or AIRE mutant mice) and thus the
polygenic nature of the disease and the small influence of most loci; 2. that
the disease results because of an immune response that once directed at a
specific beta cell peptide is essentially normal, though the targeting leads to
disease and there is spreading of autoimmunity, with implication that all
mechanisms available to the normal immune system for destruction are operative;
3. that there is a primary autoantigenic epitope presented by the class II I-Ag7
molecule and for the NOD mouse it is the insulin B:9-23 peptide presented in
“register 3” of I-Ag7 31 and recognized primarily by a conserved alpha chain (TRAV5D-4) of the T
cell receptor32 and 4. The killing of islet beta cells is asynchronous with different
islets destroyed over time until enough destruction has occurred for
hyperglycemia to develop. These “biases”
are stated, as to some extent they influence “commissions” and “omissions” of
this review that many investigators will hopefully correct in the future.

Figure 1.
Derivation of the spontaneous animal model of type 1A diabetes, the NOD mouse.
The
NOD mouse is the most-studied animal model of type 1A diabetes33-37;26, 27, 38-51. The NOD inbred mouse line was established at the Shionogi Research
Laboratories in Japan through sib breeding of a hyperglycemic female mouse from
the CTS sub line 52 (Figure 1). The line that actually became the
NOD mouse strain was originally a control line for the modestly hyperglycemic
line that became the NON strain (Non-obese Non-diabetic). NOD mice show islet
infiltration by lymphocytes around 5-7 weeks of age followed by the spontaneous
development of overt diabetes in approximately 70% of the females and 40% of
the males by 30 weeks of age 53, 54. Similar to humans, NOD mice usually (but not always) express
anti-insulin autoantibodies in their serum prior to hyperglycemia 55, 56;57 (Figure 2). Of note NOR mice which do not usually develop
diabetes follow the same age dependent pattern of expression of insulin
autoantibodies. NOR mice are a complex
congenic inbred strain with approximately 80% NOD genome and following
depletion of CD4+CD25+ T cells NOR splenocytes can mediate diabetes in NOD-scid
mice58.
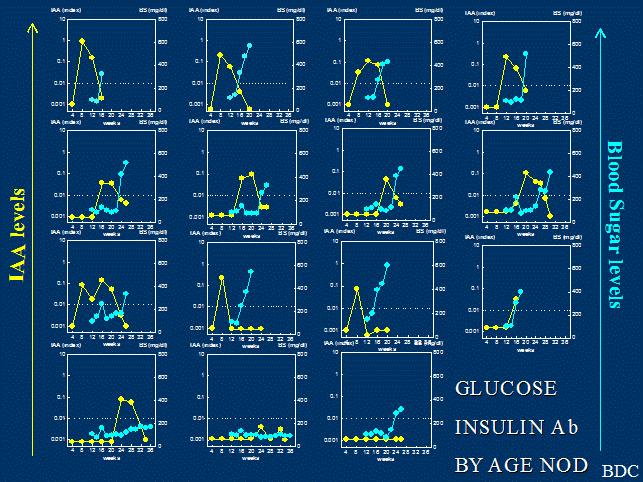
Figure 2. Insulin
autoantibodies and glucose of individual NOD mice followed from 4 weeks of age.
Although
all NOD mice develop insulitis, this is not always followed by diabetes.
Diabetes incidence varies among established NOD colonies, although in all
cases, unlike in humans, gender influences disease incidence with higher
disease frequencies in female mice than males 54. Type 1 diabetes incidence is also affected by
environmental conditions with higher disease frequencies in sterile conditions 59 and lower disease penetrance with infectious agents 60, 61. The disease can be prevented in numerous ways (>134) 26 including immunological or genetic manipulations of NOD mice 62.
Diabetes
susceptibility in the NOD mouse results from the interaction of multiple genes63, with the strongest predisposing effect deriving from genes within the
major histocompatibility complex (MHC) 64-66. Multiple studies suggest that the effect of the MHC 64 is due to the combination of the unique sequence of
the class II I-Ag7 allele with its beta chain lacking aspartic acid
at position 57 and proline at position 56 67 (the I-Aalpha chain sequence of NOD is identical to
BALB/c), a lack of expression of I-E 68 due to a common mouse mutation (also present in
C57BL/6 mice) and specific class I alleles 69, with additional polymorphisms of other genes within the MHC(e.g. idd1670, 71. In general it has been assumed
that the lack of aspartic acid at position 57 of the I-Ag7 beta
chain creating a basic I-Ag7 pocket 9 would favor the binding of
amino acids with negatively charged side chains into pocket 9. The recognition of these peptides by the CD4 T
cells then creates class II MHC mediated diabetes susceptibility. Kappler and coworkers have recently
discovered that for the insulin peptide B:9-23, just the opposite occurs. A given peptide can bind to I-Ag7
in different registers with side chains of different amino acids of the peptide
binding in pockets 1, 4, 6 and 9 depending on where the peptide docks along the
linear I-Ag7 groove (register).
T cell receptors then recognize the peptide MHC complex in a specific
register. For each specific register
that a single peptide binds in, the amino acid chains facing the T cell
receptor are different. To go from one
register to another, the peptide must rotate for specific side chains to bind
in pockets 1,4,6 and 9. By covalently coupling the B:9-23 peptide in
the groove of I-Ag7 with the arginine (peptide amino acid 22) in
pocket 9 (unfavored basic amino acid in a basic pocket) it was found that all
of the studied anti-B:9-23 autoreactive T cells reacted only when the peptide
was in this low affinity register1. This has led to the hypothesis
that such unusual recognition may allow anti-B:9-23 T cells to avoid thymic
deletion in that the concentrations of insulin in the thymus are very low. T cells should be able to recognize the
peptide in a low affinity register in the islets where the concentrations of
insulin and presumably the B:9-23 peptide presented by I-Ag7 are
huge. Of note Unanue and coworkers have
provided data that the B:9-23 peptide is likely created within islets,
potentially within insulin secretory granules with subsequent uptake by antigen
presenting cells72.
A
great deal of effort has been involved in mapping the diabetes-associated
alleles in both NOD mice and humans. In both cases, numerous loci have been
identified (>50), although to date,
few predisposing genes are identified for the NOD mouse (MHC genes 64, potentially CTLA-4 73, IL263, 74 and beta-2 microglobulin important exceptions 66, 75, 76). Genetic analysis has involved mapping of the mouse genome usually
with crossing of loci from nonautoimmune-prone strains onto the NOD background
as well as analysis of recombinant and related strains. Loci (identified as Iddn in the mouse and IDDMn in the human) with significant LOD
scores for association have been analyzed for candidate genes. Multiple
congenic strains containing Idd locus (n) from disease-resistant strains have
been generated and analyzed for disease incidence, insulitis, and immunological phenotypes (e.g., insulin
autoantibodies 77) associated with diabetes in the NOD mouse 76,
78, 79. A general finding is that several identified regions
contain multiple-disease associated loci which alone rarely confer protection
but in multitudes of two or more can nearly completely block disease 76. Positional cloning of numerous Idd loci is underway with potential for
success of identifying “causative” polymorphisms likely to be dependent upon
the number of genes within a given locus. For example a 1.52Mb region of locus
Idd5.2 contains 45 genes, while the locus Idd5.1 (2.1-Mb region) contains four
genes, including CTLA-4 63, with evidence for influence of CTLA-4 polymorphisms influencing
disease in man and mouse.
A
list of many of the multiple identified Idd loci, their positions on mouse
chromosomes, identified phenotypes, and candidate genes is presented in Table
3.1 and a more comprehensive list is provided by Serreze and coworkers 37. These loci can either confer susceptibility or
resistance to disease development and even some alleles of the NOD mouse confer
resistance rather than susceptibility. Given the identification of these loci,
it is possible to rapidly introduce loci from other strains onto the NOD
background to create what has been termed speed congenics. Usually within five
generations of backcrossing, essentially all NOD diabetogenic loci can be fixed
with the locus of interest introduced 80. Though there are reports of some success, a major
disadvantage of the NOD mouse is the lack of embryonic stem cell lines to
directly create gene knockouts on the NOD background, a lack that is partially
alleviated with the availability of “mixed” NOD strain embryonic cell lines 81.
Table 3.1 Loci of the NOD mouse (idd)
|
Locus |
Chromosome |
NOD Allele |
Phenotype protective
allele |
Disease Protection |
Genes/ (Candidates) |
Reference: |
|
idd1 |
17 |
Susceptible |
No Diabetes But some insulitis |
MHC class I and II I-Ag7 |
Hattori 1986 Todd 1988 Ikegami 2004 Serreze |
|
|
idd2 |
9 |
Susceptible |
|
|
|
Wicker 1995 McAleer 1995 |
|
idd3 |
3 |
Susceptible |
Moderate IAA, Insulitis |
69% |
Podolin 1997 Hill 2000 Lyons 2000 Ikegami 2003 Rabinow 2008 |
|
|
idd4 |
11 |
Susceptible |
age of onset |
|
|
Wicker 1995 Devidi-.. 2007 |
|
idd5 |
1 |
Susceptible |
Low IAA, Insulitis |
45% |
|
Colucci 1997 Hill 2000 |
|
idd5.1 |
1 (2.1 Mb) |
Susceptible |
CTLA-4 Ligand independent splice |
26% |
Hill 2000 Wicker 2004 |
|
|
idd5.2 |
1 (1.52 Mb) |
Susceptible |
|
0% |
Hill 2000 Wicker 2004 |
|
|
idd6 |
6 |
SusceptiblexB10 ResistantxNON |
|
|
|
Wicker 1995 McAleer 1995 |
|
idd7 |
7 |
Resistant |
Low TCR, CD8 |
|
|
Gonzlez 1997 McAleer 1995 |
|
idd8 |
14 |
Resistant |
|
|
|
Wicker 1995 |
|
idd9 |
4 |
Susceptible |
High insulitis and IAA |
90% |
Lyons 2000 |
|
|
idd9.1 |
4 |
Susceptible |
|
|
(Jak1, Lck) |
Lyons 2000 |
|
idd9.2 |
4 |
Susceptible |
|
|
(Tnfr2) |
Lyons 2000 Siegmund 2000 |
|
idd9.3 |
4 |
Susceptible |
|
20% |
|
Lyons 2000 |
|
idd10 |
3 |
Susceptible |
|
36% |
Fcgr1 ruled out, CD101 |
Podolin 1998 Ikegami 2003 |
|
idd11 |
4 |
Susceptible |
Marginal zone B cells |
62% |
|
Brodnicki 2000 |
|
idd12 |
14 |
Susceptible |
|
74% |
|
Wicker 1995 |
|
idd13 |
2 |
Susceptible |
Decrease insulitis |
100% |
β-2 microglobulin |
Serreze 1998 |
|
idd13a |
2 |
Susceptible |
|
38% |
|
Serreze 1998 |
|
idd13b |
2 |
Susceptible |
|
25% |
|
|
|
idd14 |
13 |
Susceptible |
|
|
|
McAleer 1995 |
|
idd15 |
5 |
Susceptible |
|
|
(Xmv65) |
McAleer 1995 |
|
idd16 |
17 |
Susceptible |
|
52% |
H-2k |
Ikegami 1995 |
|
idd17 |
3 |
Susceptible |
|
|
|
Podolin 1997 |
|
idd18 |
3 |
Susceptible |
|
9% |
|
Podolin 1997 |
|
Combined Congenics |
|
|
|
|
|
|
|
10/18 |
3 |
Susceptible |
Low IAA, Insulitis |
38% |
(Cfsm, Cd53, Kcna3, Rap 1a) |
Podolin 1998 Robles 2003 |
|
3/10/18 |
3 |
Susceptible |
Low IAA, Insulitis |
92% |
|
Lyons 2000 Robles 2003 |
|
3/10 |
3 |
Susceptible |
|
93% |
|
Podolin 1997 |
|
3/5.1/5.2 |
1/3 |
Susceptible |
Low IAA, Insulitis |
97% |
|
Hill 2000 Robles 2003 |
|
3/10/18/9 |
3/ 4 |
Susceptible |
|
100% |
|
Lyons 2000 |
|
10/17 |
3 |
Susceptible |
|
58% |
|
Podolin 1997 |
|
3/10/17 |
3 |
Susceptible |
|
98% |
|
Podolin 1997 |
|
5.1/5.2 |
1 |
Susceptible |
Moderate IAA, Insulitis |
50% |
|
Robles 2003 |
Table 3.1 IAA= Insulin
Autoantibodies, bold genes likely candidate.
Idd1 has been mapped to the MHC locus on mouse chromosome
17 54, 64. Although the class II allele within the MHC does not completely
explain the association of disease71, 82 to this locus (other non-MHC class II genes within this locus
contribute to disease susceptibility including class I alleles 83, 84), the MHC class II locus is the most studied genes within this region 76. The I-Ag7
molecule behaves as a recessive allele, normally required in the homozygous
state for diabetes development in the NOD model 64. Of note, both human DQ and mouse I-A
diabetes-associated sequences (DQB1*0302 and IAg7) carry a non-aspartic acid (e.g. serine at position
57 of b-chain) instead of the aspartic acid residue
conserved on other mouse strains and on other DQB1 alleles conferring low
diabetes risk in humans 65, 85. These findings have suggested that this particular residue may
influence diabetes susceptibility, but it is likely that other amino acids
within DQ or IA as well as other MHC genes may be important both in humans and
mice. In fact, population studies 86 and experiments in transgenic mice 68, 87 have failed to show position 57 alone as a susceptibility factor and in
addition support a significant role for alleles at the DRB1 locus in humans and
for the corresponding IE molecule in the mice.
The
NOD mouse lacks surface expression of the I-E molecule and its expression as a
transgene prevents diabetes 68, 87, 88. These data must be interpreted with caution since control experiments
with transgenic expression of the diabetogenic IAg7 molecule also protect NOD mice from diabetes 89. The Idd1 locus may act as a gene complex with at
least two susceptibility loci, I-A and I-E 87 as well as class I and class III loci71, 82. It must be noted that, as with any one identified Idd locus, the
combination of the I-A and I-E NOD alleles is not sufficient for diabetes
development as CTS mice share the NOD class II alleles but are
disease-resistant 90, 91. In addition, the requirement for IAg7 homozygosity is not absolute as models of mice
heterozygous for IAg7 have
been found to be susceptible to type 1 diabetes 92.
Recent
crystallography studies of the IAg7 molecule showed this MHC protein is structurally
stable. In comparison to other class II molecules, this diabetes-associated
allele contains an altered peptide binding groove which is reported to allow
more promiscuous binding of numerous peptides 93, 94, potentially related to earlier studies of weak peptide binding 95. Furthermore, diabetes-associated DQ alleles in
humans were also found to have similarly altered peptide-binding grooves in
crystallography studies (31). One possible role of weak presentation of self-peptides
by MHC in autoimmunity could be inefficient thymic deletion. Support for this
concept comes from limiting dilution studies which showed a high frequency of
autoreactive T cells in IAg7 mice 96. In general though it appears that I-Ag7 functions normally
in peptide presentation and has high affinity for a subset of peptides. The hypothesis that there is some overarching
abnormality of the class II molecules associated with autoimmunity is difficult
to reconcile with the observation that some MHC haplotypes protect from one
autoimmune disease, while enhancing another, such as the DRB1*1501, DQB1*0602
haplotype association with human multiple sclerosis and type 1 diabetes 97.
Investigations
into the role of MHC on thymic tolerance processes provide evidence for both a
lack of thymic deletion in IAg7 homozygous mice and positive selection of regulatory
T cells by protective MHC alleles. The CD4+ anti-islet 4.1 TCR transgenic is positively selected
by IAg7 but
negatively selected with coexpression of other MHC class II alleles 98. In addition, Ridgway and coworkers showed that
dosing the IAg7 allele
correlates with the degree of insulitis and autoreactive repertoire 99. On the other hand, BDC2.5 TCR CD4+ anti-islet transgenic mice show selection of
regulatory subsets by protective alleles 100. Thus, the current data suggest altered MHC alleles
result in inefficient thymic deletion and altered positive selection in the NOD
mouse. However, there is also evidence for a role of the IAg7 molecule in the peripheral activation of
autoreactive T cells 101. A mutated form of the IAg7 molecule replacing the His and Ser at positions 56
and 57 with the more common Pro and Asp residues generates a transgenic MHC
molecule known as BALBg7PD. This MHC transgenic mouse mediates positive
selection of BDC2.5 TCR transgenic cells, however, these mature T cells are
unable to mount an anti-islet response with BALBg7PD antigen-presenting cells
(APCs) whereas responses with NOD WT APCs are adequate 102. In conclusion, the Idd1 MHC locus may contribute to type 1 diabetes
both in altering thymic selection processes and facilitating activation of
autoreactive T cells in the periphery. Several groups including our own have
provided evidence that insulin may a primary autoantigen for the NOD mouse and
in particular insulin peptide B:9-234, 5, 103-105. If this insulin peptide is
indeed essential for the development of diabetes of the NOD mouse, the manner
in which the B:9-23 peptide binds to I-Ag7 may be a crucial
determinant of disease, with report of two binding registers (register 1 and 2)105. Though a crystal structure of the complete
trimolecular complex of I-A87 – B:9-23 peptide and relevant
anti-B:9-23 receptor is needed, John Kappler’s laboratory has provided evidence
that the peptide binds to I-A87 in a very low affinity register
(register 3) in terms of TCR recognition1. The B:9-23 peptide appears to
primarily be targeted by a germ-line encoded V alpha chain (TRAV5D-4) with
marked variation in TCR alpha CDR3 and multiple TCR beta chain sequences 106, 107.
The
BDC 2.5 target antigen is a natural endocrine cell processed peptide of chromagranin,
WE-14. “Remarkably” the exact site of N-terminal cleavage to produce WE-14 is
essential for BDC 2.5 T cell receptor recognition and the I-A87
groove is only partially filled by the WE-14 peptide 2. A recent manuscript suggests
there is an additional totally different chromogranin peptide also reactive
with the BDC 2.5 T cell receptor but this study did not yet study clonal BDC
2.5 T cells. 108.
Idd2 has been assigned to mouse chromosome 9 and linked
to the T lymphocyte marker thy1, although a significant association with
diabetes has not been found in all studies 109.
Idd3
Progress in gene identification and
contribution to disease has been made with the Idd3 locus with major candidate
genes encoding cytokines IL2 and IL21 with at present no clear distinction
between the two candidates 37. NOD mice congenic for the Idd3 region of chromosome
3 from C57BL/6 mice show a low incidence of diabetes (25% compared to 80%) 110. The Idd3 locus contains the IL2 gene which has a unique glycosylation
form in the NOD 111, however, the protein appears to have normal function and the
glycosylation difference has been genetically ruled out as contributing to
diabetes74. Identified idd3 candidate genes
in what is now approximately a 650Kb congenic region include Tenr, IL2, IL21, and Fgf2, and Cetn4, 83 76 and two genes of unknown function63, 112. There is a lower expression of IL2 and a higher expression of IL21
with the risk NOD locus. A combination of idd1 and idd3 introgressed onto the
C57BL/6 strain is not sufficient for the induction of diabetes with C57
background genes 113. Engineered haploinsufficiency
of IL2 similar to low expression of IL2
of the NOD mouse associated with idd3114 locus results in reduced CD4+CD25+ regulatory T cells and enhanced
diabetes74.
Idd4
maps to chromosome 11 and may influence the frequency and severity of insulitis
and progression to diabetes 54, 59. In particular, Idd4 homozygosity determines the age of onset of
diabetes and Idd2, Idd3, and Idd4 together may accelerate progression to overt
diabetes 115. A recent report indicates that NOD mice with
deleted lipoxygenase involved in the
production of proinflammatory fatty acids increase the development of diabetes116. In addition constituitive phosphorylation of Stat5 of NOD mice is
associated with idd4117.
The
Idd5 locus is located on chromosome 1 and alone confers 50% protection
from diabetes and in combination with Idd3 provides nearly complete protection
from infiltration of the pancreas, thyroid, and salivary glands 118. Therefore, these genes are likely to influence
tolerance processes in the animal 119. The synergistic effect follows a model of additivity rather than
multiple epistasis 120. Two loci have been identified within the Idd5 region which results in
delayed onset of disease. Idd5.1 overlaps with the IDDM12 locus in humans
(candidate gene is CD152 or CTLA-4) 118 and which has been narrowed to 2.1-Mb containing CTLA-4,
ICOS, Als2cr19, and Nrp2, with CTLA-4 being the primary candidate 73, 121 but higher levels on T cells of ICOS on NOD T cells 122. The susceptible idd5.1 allele
is associated with low levels of a CTLA-4 splice variant that lacks the ligand
binding domain to CD80/86 123. Idd5 F2 mice
show a resistance to gamma-induced apoptosis in the NOD and NOR strains while T
cells from C57BL/6 and DBA/2 strains show a “high-apoptosis” phenotype 124. The CTLA-4 candidate gene is intriguing because CTLA-4-/- mice are
also resistant to apoptosis 125. The NOD idd5 locus mediates a bone marrow cell derived defect in
negative selection of t cells126. Microarray data have implicated
CD55 (decay accelerating factor) and acyl coenzyme A dehydrogenase expression127 associated with idd5.
The
Idd6 locus contains the NK cell cluster; NOD.Idd6 (containing NK1.1)
congenic mice have been shown to have reduced disease incidence 128. The importance of NK cell function in reducing disease in NOD mice has
been a story of great interest 128-130, although the complete localization to Idd6 is still
unknown. F2 intercrosses
between NOD and C57BL/6 mice showed an association between the Idd6 locus on
chromosome 6 and decreased proliferation of immature thymocytes in NOD mouse 131, although the contribution of this phenotype to disease is still
unknown. Additional phenotypes associated with idd6 loci include downregulation
of expression of Toll like receptor 1 and decreased expression of the Lmp gene97, 132.
A
recent study indicates that the IDD7
locus influences thymic deletion of specific CD8 autoreactive T cells such as
AI4133 and this relates to low expression of the chain of the T cell receptor
of AI4.CD8 T cells 133. Little information is available
on Idd8 except that this locus appears to be protective in the
homozygous state 115.
The
Idd9 locus on chromosome 4 is now known to contain 3 distinct loci.
NOD.B10 Idd9.1/9.2/9.3 triple congenic mice are almost completely protected
from diabetes yet still show insulitis, suggesting these loci are involved in
regulating autoreactive T cells 110. A change in cytokine production from IFNg to IL4 by infiltrating cells in triple congenic mice
compared to wild-type NOD supports this interpretation. The presence of
insulitis and salivary gland infiltrates in the Idd9 congenic mice suggests
that tolerance defects still exist. Genes of the TNFR superfamily with
polymorphisms between NOD and B10 mice include candidate genes CD30 and TNFR2
for Idd9.2 134 and CD137 for Idd9.3 110.
The
idd9.1 locus is associated with greater development of NK T cells which may
promote immunoregulation 135. Idd11 has been localized to overlap with the Idd9.1 locus.
It is reported that in Idd9 mice autoreactive T cell accumulate in the
pancreatic lymph node133 and has been reported to influence marginal zone B cells but not
confirmed with congenics 136, 137.
Idd10 and Idd18 are closely linked on mouse
chromosome 3. NOD.B610/18 congenic mice show reduced incidence of diabetes (50%
vs. 80%) and include candidate genes Csfm, CD53, KCNA3, Nras, and Rap1a 138. Recent studies evaluating the IIS mouse139 and with congenic mapping indicates that Idd10 is not Fcgr1 83 and CD101 that differs in sequence from NOD for IIS and B6 is a
candidate. Adding another locus from chromosome 3 to create NOD.B6 3/10/18
congenic mice results in almost complete loss of diabetes as well as insulitis 140. The few animals that do develop diabetes have a delayed-onset.
Idd13, like idd5, is involved in the regulation of T cell
progression from benign to destructive insulitis. It contains at least two
identified loci: idd13a and idd13b, which contains the gene b2-microglobulin 79. The studies of Serreze and coworkers have firmly
established a polymorphism of b2-microglobulin as influencing development of NOD
diabetes, and it is one of the few established “genes” outside of the MHC 75. The NOD allele is a standard a isoform, and the b
isoform differs from the a by one amino acid (alanine instead of
aspartic acid at amino acid 85). The b allele does not suppress diabetes
in the presence of the a allele, but the b allele cannot restore
diabetes development by transgenesis in mice lacking beta-2 microglobulin but
the a isoform does 75. NOR derived idd13 locus
increases invariant NKT cells141.
“Monogenic
Autoimmune Mutations”
In
1926, Schmidt described a patient with Addison’s disease and thyroiditis 142, and eventually several clinical syndromes
consisting of multiple autoimmune disorders were recognized 13. A subset of these syndromes develops from single
gene mutations; many such single gene mutations now have animal models. In
particular, the Autoimmune Polyendocrine Syndrome Type 1 (APS-I) results from a
mutation of the AIRE gene and approximately 18% of patients with this syndrome
eventually develop type 1 diabetes (usually with Addison’s disease,
mucocutaneous candidiasis, and hypoparathyroidism) 13. When this mutation(deletion) is bred onto mouse
strains, lymphocytic infiltrates occur, but no diabetes 24, 143, 144. Of note, however, the AIRE mutation appears to have a major role in
expression of “peripheral antigens” within the thymus 23. Hanahan coined the term “peripheral” antigens 145-147, as molecules expressed at low levels within the thymus (e.g.,
rat-insulin-promoter-driven expression in his studies) and it is clear that
such expression has a major influence on autoimmunity to the respective
molecules. There is one Italian family reported with an autosomal dominant form
of APS-I148 and Anderson and coworkers have introduced this mutation into NOD mice6. An autosomal dominant
autoimmune syndrome develops that differs from the phenotype of NOD mice with
both AIRE genes knocked out. In
particular the autosomal dominant disease does not result in pancreatitits6. The dominant negative mutation
appears to act by recruiting wild type AIRE away from active sites of
transcription and does down regulate expression of peripheral antigens within
the thymus. The AIRE knockout on NOD mice did not accelerate the development of
diabetes and combined myd88 knockouts (eliminating major pathways of toll like
receptor signaling) or raising mice in a germ free environment did not
influence disease149.
The
IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) syndrome
is another particularly informative autoimmune syndrome, with mutations of the Foxp3 gene and the homologous scurfin gene in mice. This mutation results in loss of a
major subset of regulatory T lymphocytes (CD4+CD25+), and
overwhelming autoimmunity in man, such that children often die as neonates, and
immune-mediated diabetes can occur in the first days of life 22, 150. In the scurfy mouse, the disease can be cured with partial T cell
chimerism, and the same appears to be true of man with normal T lymphocytes
able to regulate the abnormal immune system in a dominant fashion 151. Mice with
the foxP3 (scurfy) mutation die from overwhelming autoimmunity, but T cell
receptor transgenics combined with this mutation, allow studies of specific
autoimmunity, and accelerated development of diabetes152. Depending upon the specific T
cell receptor, the Fox P3 mutations can
accelerate diabetes development even in
rag-/- mice expressing a single anti-insulin B:9-23 T cell receptor (Jasinski
et al unpublished).
The more common
autoimmune disorders, similar to the NOD mouse, are polygenic in origin. Human
and rat studies of type 1 diabetes, multiple sclerosis, collagen-induced
arthritis and systemic lupus erythematosus (SLE) have been mapped to the same
chromosomal sites, suggesting a common genetic basis 78, 153, 154. However, given the number of loci, some chance overlap is to be expected.
The autoimmune polymorphism of the
PTPN22 gene (LYP gene of man) contributes
to multiple autoimmune diseases (type 1 diabetes, rheumatoid arthritis, Graves’
disease, lupus erythematosus) 155-157). The NOD mouse itself develops multiple autoimmune manifestations in addition
to type 1 diabetes including thyroiditis, lymphocyte infiltration of the
salivary and lachrymal glands 66, and can develop polyneuropathy 158 if B7.2 deficient. NOD mice have
been reported to target “Schwann” like cells surrounding islets 159, and are susceptible to both experimental autoimmune encephalitis (EAE
- an animal model for multiple sclerosis) 160 and experimental autoimmune prostatitis 161. Therefore certain Idd genes or combinations thereof alter the immune system such that tolerance
processes are defective for multiple tissues. The form of autoimmunity may then
depend on other genetic (e.g., MHC and antigens) or environmental factors. One
recent study shows that autoimmunity in the NOD switches from pancreas-specific
to destruction of the peripheral nervous system by altering costimulatory molecules present on dendritic
cells 158 and NOD.c3c4 mice with multiple B6 and B10 protective alleles from
chromosome 3 and 4 bred onto NOD do not develop diabetes, but develop
autoimmune biliary disease 162. Of note, breeding only one of the two chromosomal regions onto NOD
produces mice with biliary autoantibodies without liver infiltration 162. In a similar manner, NOD congenics can express anti-insulin
autoantibodies with rare progression to diabetes 77. Thus disease targeting is influenced both by MHC
alleles and by other polymorphisms in an autoimmune “background”.
Immunology of the NOD mouse
Beta cell destruction in the NOD requires CD4 T cells,
CD8 T cells 163-165 and B cells for its spontaneous occurrence 166, and maternal transfer of autoantibodies has also
been found to have a role 167.
Th17
cells do not apprear critical for initiation of insulitis168. The NOD mouse allows studies of
the progression of the disease including the timing of insulitis and the nature
of insulitis, i.e., cytokine production during the course of the disease. Prior
to developing sufficient beta cell damage to produce hyperglycemia, a period of
insulitis is observed followed by eventual development of hyperglycemia. There
remains a debate among investigators whether the beta-cell loss is a gradual or
acute process, occurring suddenly once regulation of autoreactive T cells
wanes, but we suspect the term “benign” insulitis is a misnomer. More than 90%
of untreated female NOD mice lose beta cell mass with time, such that by 52
weeks beta cell mass is 1/5th to 1/10th of normal even
for the great majority of female mice that have not progressed to diabetes 169 (and Gianani et al ADA abstract 2008). There is
considerable endocrine reserve and mice even with 1/5th of normal
beta cell mass can remain non-diabetic. As illustrated in Figure 3, even 8-9
week old NOD mice have lost beta cell mass. Of note, the histology of the NOD
pancreas suggests asynchronous destruction of islet beta cells. Within the same
mouse, one can find normal islets with beta cells, islets with insulitis and
beta cells, and islets where all the beta cells (insulin producing cells) have
been destroyed, and only non-beta cells (e.g., alpha cells producing glucagon)
remain. Additionally during the phase of active destruction there is evidence
of beta cell replication 169, 170 that eventually cannot keep pace with beta cell destruction. Recent three dimension analysis of beta cell
mass over time indicates that beta cells on the periphery of the pancreas are
destroyed first with increased size of central islets, followed by their
destruction 171.
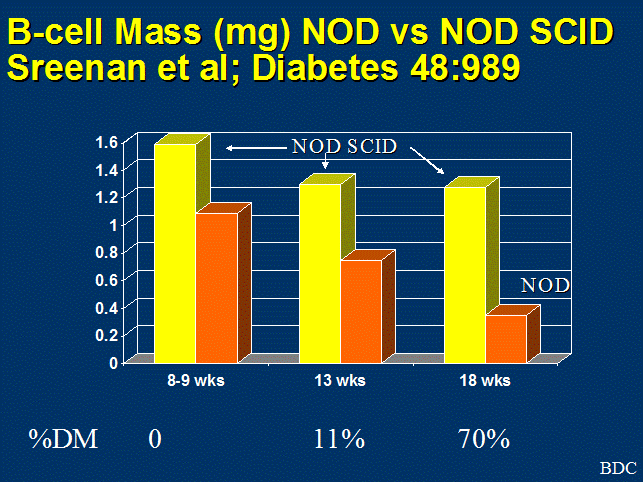
Figure 3. Quantitation of beta cell mass, comparing
NOD-SCID mice that lack T cells, to loss of beta cell mass in NOD with age.
There is now clear
evidence that mice can regenerate beta cell mass following acute beta cell
destruction but it is not clear if humans have such an ability40, 172. Anecdotal reports of beta cell
proliferation173 followed by analysis of multiple individuals174 suggest that replication of human beta cells is infrequent in adults 175. A subset of patients with type
1 diabetes retain C-peptide secretion long-term and infrequent scattered beta
cells are present in most long-term patients 176, but the great majority have very low c-peptide levels176. The mechanism related to
retention of some beta cell mass in man and in the NOD mouse model is currently
unknown. Herold and coworkers analyzing
NOD mice prior to the development of diabetes and then during therapy with both
anti-CD3 and regulatory T lymphocytes have concluded that inflammation
increases beta cell replication, that most of the recovery of beta cells
following therapy is a result of regranulation of degranulated beta cells and
following therapy beta cell replication is reduced177 with lower percentage of Ki67+ beta cells post recovery(figure below).
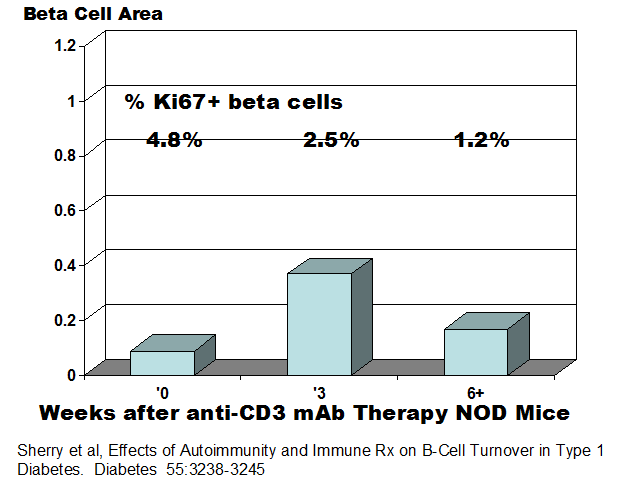
In NOD mice,
expression of insulin autoantibodies (IAA) is usually noted prior to onset of
hyperglycemia 178. In addition, the NOR mice, a strain closely related
to the NOD with the IAg7 MHC
haplotype as well as other NOD congenic strains 30, 77, develop IAA but with limited or no progression to type 1 diabetes 77. The presence of insulin autoantibodies correlates
primarily with insulitis and not simply progression to hyperglycemia. Genetic
studies using congenic NOD strains show the expression of IAA antibodies is
controlled by multiple loci. 77 In addition, the amount of IAA was found to
associate with degrees of insulitis more than disease incidence 77. There is
evidence of additional autoantibodies reacting with islets cell antigens of NOD
mice, but in workshops it has been difficult to confirm the presence of such
autoantibodies with highly specific fluid phase radioassays179, 180. It is likely that B-lymphocytes
(though insufficient by themselves181 can not only contribute to diabetes through the production of
autoantibodies (e.g. evidence that “transplacental” passage of autoantibodies
of NOD mice important for disease182) but have additional roles, particularly as antigen presenting cells
and for the maturation of CD8 T cells167, 183. Treatment of “humanized” CD20 NOD mice by an anti-CD20
monoclonal in clinical use prevents the bulk of diabetes184.
It
is likely that antigen presentation for the activation of pathogenic T
lymphocytes occurs in the pancreatic lymph node with evidence provided by the
BDC 2.5 transgenic T cells, where labeling of cells with the dye CFSE indicates
initial proliferation in such lymph nodes 185, 186, the finding by Fathman and coworkers that pathogenic cells in the
pancreatic lymph node are CD4high 187, and removal of pancreatic lymph nodes at 3 weeks but not 10 weeks
prevents diabetes 188. The role of various cell populations in the beta cell damage have been
addressed using knockout animals, T cell clones, and adoptive transfer models
of the NOD mouse and are discussed in the following chapter. To summarize
numerous findings, spontaneous disease requires CD4 and CD8 T cells as well as
B cells, which are thought to be involved in antigen presentation. Mathis and
colleagues have implicated NK specific transcripts and proportion of NK cells
in development of destructive islet autoimmunity 189.
The
confirmation that lymphocytes are required for the beta cell destruction in
type 1 diabetes led to numerous studies on the immune system in NOD mice
compared to nonautoimmune-prone strains. A deficiency in the ability of NOD
APCs to mount equivalent T cell responses has been reported 190, 191. These NOD APCs have been found to have low CD86 expression 192, and recent studies have reported phenotypic and functional defects in
bone-marrow-derived dendritic cells (DCs) in response to GM-CSF 193. Wong et al. have found a deficiency in priming of NOD T cell responses
to both endogenous and exogenous antigens 194. Similarly, defects in TCR-mediated signaling
resulting in inferior NOD T cell responses have been linked to alterations in
p21ras signaling 195. Neonatal CD28 costimulation has been recently found to restore this
signaling as well as protect NOD mice from diabetes, suggesting a relationship
between this T cell hyporesponsiveness and disease onset 196. These abnormalities in T cell function may relate “lymphopenia” of the
NOD mouse with the hypothesis that homeostatic expansion may contribute to
autoimmunity 197.
Bellgrau
and coworkers found a similar T cell hyporesponsiveness among numerous strains
of autoimmune-prone mice susceptible to EAE, SLE, rheumatoid arthritis, and autoimmune
hemolytic anemia 198. The prevention of diabetes resulting from administration of immune
adjuvants 199, 200 or DNA vaccinations to self-antigens 201 may be related to reversing this hyporesponsive immunity observed in
the NOD. Further work is required to clarify the mechanism of protection and
possible role of a hyporeactive immune response in autoimmunity.
One
possible mechanism by which a hyporeactive T cell response may contribute to
autoimmunity is through defective negative selection in the thymus. Evidence
for poor central tolerance in the NOD includes direct demonstration of inferior
thymic deletion in NOD compared to BALB/c mice upon administration of anti-TCR
antibodies, increased frequency of autoreactive T cell responses to pancreatic
antigens 96, 202-205, and demonstration that epitope
spreading in NOD follows a hierarchy from highest affinity TCR:self-antigen
interactions to lower affinities suggesting the spreading hierarchy is
determined by the extent of negative selection 206. The concept that with disease progression lower affinity TCR reactions
are favored is challenged by the studies of Santamaria and coworkers who
observe affinity maturation of the TCR utilized by cells targeting the molecule
IGRP (islet-specific glucose-6-phosphatase catalytic subunit-related protein),
which is the native molecule targeted by anti-NRP (NOD Related Peptide) CD8
lymphocytes which form the majority of T cells infiltrating islets 207-209. Other thymic defects in the NOD include abnormal corticomedullary
environments 210, a deficiency in the number of TCR+ CD4-, CD8- populations in NOD mice, and low proliferation of
immature thymocytes 131. Whether these phenotypes have relevance to autoimmunity remains to be
determined, although the deficiency in TCR+ DN thymocytes does not appear to be related as it is
seen in a number of congenic Idd mouse strains which are not diabetic 211.
Regulatory Cells
Table 3.2: Summary of Regulatory Cells influencing
diabetes
|
Regulatory
Cell |
Induction |
Comment |
Reference |
|
CD4+ TGFbeta |
Insulin oral |
Oral insulin delay of
diabetes in NOD mice. Did not delay BB rat diabetes |
Zhang (1991) |
|
CD4+CD62L+ |
Anti-CD3 |
Anti-CD3 treatment
induction, TGFbeta dependent mechanism |
Belghith (2003) |
|
CD4+CD25+ |
BDC2.5 |
TCR Clonal and Mimotope
driven delay/prevent diabetes CD28-/- loss regulation,
greater diabetes |
Tang (2004) |
|
(CD69+, CD45RBintCD62Lhigh) |
Insulin B:9-23 |
Specific 100% prevention
diabetes transfer into SCID, in vivo and in vitro B:9-23;
Secrete TGF-β, TNF-α |
Mukkherjee (2003) |
|
NK T cells |
GalCer |
Experimental activation
with GalCer protects |
Sharif (2001) |
|
γδ T cells |
Administration of insulin
by naso-respiratory route (“no degradation”) induces IL10 producing |
|
|
|
NK cells CD3-DX5+ cells |
|
Complete Freund’s adjuvant
protection |
Lee (2004) |
|
Bitypic NK/DC Regulatory
cells |
Anti-CD40 ligand |
Prevent diabetes in
RIP-LCMV model |
Homann (2002) |
|
GAD
Transgenic |
Anti-GAD
TCR |
Two
anti-GAD TCR transgenics prevent NOD diabetes |
McDevitt
(2004) |
It
is well established that autoreactive T cells can be controlled by regulatory
cells (Table 3.2). There has been a recent tremendous expansion in
knowledge concerning regulatory T lymphocytes 11, 212, and in particular, their role in the NOD mouse and the ability to
generate large numbers of CD4+CD25+ regulatory T cells213, 214 and prevent diabetes 212. Evidence for the role of regulatory T cells in delaying or preventing
overt diabetes are numerous including prevention of adoptive transfer of
diabetes by mixing splenocytes 215 from pre-diabetic NOD mice with the disease-inducing splenocytes from
diabetic donors 216, induction of regulatory cells dependent upon TGFbeta with anti-CD3
therapy in recent onset NOD mice 217, and the large literature concerning mucosal tolerance with bystander
suppression, including ability of oral insulin to delay diabetes of NOD mice 218. It is clear that these regulatory T cells are heterogeneous in nature,
and distinct populations have been elucidated 47, 213, 219-222.
Related
to the initial studies of thymectomy-induced autoimmunity 223, 224, the remarkable human autoimmune phenotype of mutations of the Foxp3
gene (IPEX syndrome-see chapter 8) associated with neonatal immune mediated
diabetes 150, the ability of Foxp3 expression to generate CD4+CD25+
regulatory T cells 225 and suppress NOD diabetes 226, and the elegant models inducing deficiencies of these regulatory T
cells 223, 227, the CD4+CD25+ T cells have become perhaps the
best characterized regulatory T cell of the NOD mouse 11, 212, 228, 229. NOD mice genetically deficient for CD28 expression succumb to diabetes
with faster kinetics than wild type NOD mice 230. This accelerated disease incidence was found to be
associated with a loss of CD4+CD25+ regulatory T cells.
Of note, as published by Bluestone and
coworkers, 107 in vitro expanded FoxP3 expressing Tregs
(using the BDC2.5 T cell receptor transgenic (recognizes islet membrane
antigen) as proof of principal) reversed new-onset diabetes in 60% of NOD mice.
BDC2.5 Treg cells have been demonstrated in the pancreatic islets and may act
at this site to suppress disease given the ability of ICOS blockade to
accelerate development of diabetes 231. Similar regulatory T cells have been induced by immunization with the
B:9-23 insulin peptide as well as a B24-C36 proinsulin peptide 232, 233.
A
critical role of NK T cells in protection from diabetes has recently been
demonstrated in NOD mice. A deficiency in number and function (IL4 secretion)
of CD1-restricted NK T cells has been observed for many years 234, 235, however, the contribution of this phenotype to diabetes development
was unclear. Several recent studies demonstrate the protection offered by
increased numbers or function of CD1-restricted NK T cells 128-130. NK T cells show a restricted
TCR repertoire with a predominance of Va14/Ja281 chains. These cells are specific for lipid
antigens presented in the CD1 MHC molecule. Upon activation, which can be
achieved with alpha-galactosylceramide (aGalCer), NK T cells produce large amounts of
cytokines, most notably IL-4 and IL-10. The Idd6 locus contains the NKR-P1 gene
cluster, which includes the NK1.1 gene. The NK1.1 gene is absent in NOD mice,
making NK T cells more difficult to analyze. Carnaud and coworkers demonstrated
a contribution of the Idd6 locus to disease incidence as NOD.NK1.1 congenic
mice have reduced diabetes incidence which correlated with improved NK cell
function 128. The contribution of NK T cell defects to diabetes incidence was also
demonstrated in NOD CD1 KO mice which showed an exacerbated disease course. In
addition, increasing the numbers of NK T cells in the NOD by transgenic
expression of the Va14Ja28 receptor resulted in reduced disease 129. The protection appears to be related to increased IL4 production
observed in the pancreas as IL12 or anti-IL4 antibodies were shown to abolish
this protection 129. It appears that increasing numbers of NK T cells is not necessary for
protection as activation of the existing NK T cells with GalCer alone
protects diabetes in CD1-sufficient NOD (50-52) 130 but not in
CD1-deficient NOD mice (52). This CD1-restricted NK T activating reagent also
enhances the survival of transplanted islets 130.
NOD
mice injected with complete Freund’s adjuvant are dramatically protected from
the development of diabetes. Lee and coworkers have recently reported that this
protection is abrogated when NK asialo-GM1 cells are depleted and restoring
cells expressing CD3-DX5+ restored protection 236.
Harrison
and coworkers have studied the administration of insulin by various routes as a
means of protecting NOD mice. They find that administration of insulin by the
naso-respiratory route protects from diabetes with the induction of CD8+
alpha (TCR gamma delta T regulatory cells that can prevent NOD diabetes). These
cells cannot be generated in day 3 thymectomized mice, but are restored with
administration of the above gamma delta T cells 233.
Singh
and coworkers have reported that insulin B:9-23 peptide immunization generates
CD4+CD5+ regulatory T cells capable of preventing
diabetes 232
Homann
and coworkers have utilized anti-CD40 ligand blockade and the RIP-LCMV diabetes
model to generate cells with the unusual set of markers of both NK cells and
dendritic cells, with the finding that these cells can block the development of
diabetes 237.
Antigenic Targets
A
fundamental question relative to the immunopathogenesis of type 1A diabetes
(immune- mediated diabetes) is whether there are primary islet autoantigens. It
is clear that multiple islet molecules 51 are the target of autoimmunity in man and animal models 238, 239. In particular, there are T cell clones 240 whose target antigens are currently unknown and a list of well-characterized “specific” targets for T cell clones (e.g.,
insulin, IGRP, proinsulin, chromagranin) and newly described islet targets
(e.g. Pdx- 1 Pancreatic duodenal homeobox) 241as well as target molecules such as heat shock proteins, GAD, dystrophia myotonica kinase (DMK) regenerating
gene II 242,where the antigen is either minimally or not specifically expressed in
mouse islets (GAD) or neurendocrine cells243 or widely expressed in multiple tissues 244, 245.
In
terms of islet autoantibodies of NOD mice, two autoantibody workshops suggest
that only insulin autoantibodies can be specifically detected using sensitive
radioassays 180, 246. Despite the large number of islet autoantigens, there is increasing
evidence that insulin, and in particular, a specific epitope of insulin (the
Wegmann insulin B chain amino acids 9-23) may be an essential target in the NOD
mouse model 30, 232, 247-249. Both cloned CD4 and CD8 T cells reacting with an overlapping insulin B
chain peptide can mediate adoptive transfer of type 1 diabetes 194, 238. MacLaren and coworkers reported that subcutaneous administration of
insulin and insulin B chain prevents the development of diabetes in NOD mice,
and Weiner and coworkers reported that oral insulin decreased the development
of diabetes 250-253. Wegmann and coworkers isolated T lymphocytes directly from islets of
NOD mice. The T lymphocytes were stimulated with islets as “antigen” and
following the development of a series of clones, the majority was found to
react with insulin 238, 247. Of the clones reacting with insulin, more than 90% reacted with
insulin peptide B:9-23. These clones were notable for utilizing conserved T cell receptor V alpha [TRAV5D-4 (AV13S3)]
J alpha gene and AJ53 T cell receptor segments, with variation in the
junctional region and no apparent conservation of the Vb chain 254, 255. Despite utilization of this dominant Va-chain motif, one of the clones studied recognized
insulin peptide B:9-16 and another four B:13-23 256. Administration of the B:9-23 peptide either
intranasally without adjuvant or with a single injection in incomplete Freund’s
adjuvant protects the majority of NOD mice from progression to diabetes 257. Wong and coworkers have isolated a NOD CD8 clone
reacting with insulin peptide B:15-23 and reported a very high frequency of
B:15-23 tetramer-positive cells within islets of NOD mice, though the exact
percentage remains controversial 194, 258. Follow-up studies suggest that though present early in lesions the
percentage of CD8 anti-B:15-23 T cell clones is more limited with a larger
population of IGRP (islet-specific glucose-6-phosphatase catalytic
subunit-related protein)-reactive CD8 T cells 239. Santameria and coworkers have immunized NOD mice with peptides of IGRP
on nanoparticles and induced high affinity CD8+ T cells that reverse
diabetes 259.
Autoantigen Gene Knockouts and Retrogenics
Baekkeskov
has produced a GAD65 knockout and breeding this gene onto NOD mice does not
effect diabetes development.260 Similar studies of knocking out potential islet
target molecules such as IA-2 and IA-2 beta similarly did not influence
progression to diabetes261, 262. Lack of immune response to
GAD65 did not influence progression to diabetes243, 263 but there is interesting evidence that anti-GAD responses can be
protective263.
Vignali
and coworkers have produced a series of retrogenics (bone marrow transplants of
stem cells with T cell receptors) with T cell receptors targeting islet
molecules. Though anti-GAD T Cell
Receptors did not induce disease, anti-insulin (BDC12-4.1T cell receptor)
induced delayed diabetes264 (Note anti B:9-23 12-4.4 TCR sequence studied in that manuscript was
different from our diabetogenic BDC12-4.4 retrogenics.). Further studies by Vignali of a large series
of anti-GAD TCR retrogenics documented induction of encephalitits and high
titer GAD autoantibodies but no insulitis or diabetes 265. Vignali’s anti-GAD T cell
receptor retrogenics included 10E1 that target GAD peptide 524-543, the same
target as the 5A anti-GAD CD4 T cell line that, on transfer into SCID mice, caused diabetes 266but not insulitis of diabetes.
Mouse islets contain almost no GAD and insulitis can be induced in
anti-GAD TCR retrogenics in mice only if induced to express GAD in beta cells
(GAD-transgenic) 260. This
suggests that presence of GAD65 is irrelevant to the spontaneous development
Table 3.3 Autoantigen Knockouts
|
Knockout |
Insulin Autoantibodies |
Insulitis |
Diabetes |
Reference |
|
Insulin 1 |
Unaffected |
Modest Decrease |
90% Prevention |
Moriyama 2003 |
|
Insulin 2 |
Increased |
Increased |
Accelerated |
Moriyama 2003 Boitard 2003 |
|
GAD65 |
|
|
No Effect |
Kash 1999 Yamamoto 2004 |
|
IA-2 |
|
|
No Effect |
Kubosaki 2004 |
|
IA-2beta (Phogrin) |
|
|
No Effect |
Kubosaki 2004 |
of diabetes of NOD mice but
does not address a potential role of GAD67 and the ability of GAD peptide
immunization to prevent diabetes. Yoon and coworkers produced five anti-sense
GAD transgenic lines 267 with the transgene bred onto the NOD background. Follow-up of these
lines indicates that of the two lines with any diabetes suppression, one
develops diabetes spontaneously and the other after cyclophosphamide induction
(oral communication). Further study of these transgenic lines will be of
particular interest relative to the mechanism of disease alteration.
The
lack of effect of the GAD65 knockout is concordant with studies inducing
tolerance to GAD and finding no influence on progression to diabetes 243. Nevertheless anti-GAD T cell
receptor transgenes mice inhibit
development of diabetes 263. For the NOD mouse there is a consensus that immune recognition of GAD
is more related to protection than beta cell destruction263. In addition, at least one human
DR4-restricted anti-GAD TCR transgenic targeting islets has been produced that
causes insulitis, but not diabetes 268.
HSP60
and peptide 277 have not been studied with knockout techniques. Cohen and
coworkers have studied in detail the ubiquitous HSP60 molecule and peptide
p277-responsive T lymphocytes of NOD mice 269-272. The administration of the p277 peptide has been reported to prevent
diabetes, but to have no effect in a study by Atkinson and coworkers 273. The evidence that HSP60/p277 is an islet
“autoantigen” is relatively weak, with no demonstration in workshops of
specific reactivity and with an alternative hypothesis for its effects in NOD
mice related to activation of the innate immune system 245.
Insulin as the Primary Autoantigen of the
NOD Mouse
To date, only knockouts of
insulin genes have influenced development of NOD diabetes (Table 3.3). Mice
have two insulin genes, and since both insulins are present in islets and both
insulins are metabolically active, it is possible to knock out either gene and
not develop metabolic forms of diabetes. The insulin 2 gene is the proinsulin
gene that is expressed within the thymus. The proinsulin 1 gene is a
retroposon, with almost no expression within the thymus. Single insulin 2 gene
knockouts (produced by J. Jami) were bred onto NOD mice by our group 30 and Boitard and coworkers 104, 274. The insulin 2 gene knockout greatly accelerates the development of
diabetes and increases the levels of insulin autoantibodies. The insulin 1 gene
knockout prevents approximately 90% of the development of diabetes of female
NOD mice but does not alter the expression of insulin autoantibodies and the
majority of mice with the insulin 2 knockout have insulitis, consistent with
the subset progressing to overt diabetes. Jaeckel and coworkers using a
technology to induce tolerance similar to that which failed to alter
progression to diabetes with GAD65 transgene 243, have recently reported that with an insulin 2 preproinsulin transgene
with invariant chain promoter, progression to diabetes is markedly decreased.
They concluded that insulin is a “key” autoantigen of the NOD mouse but not
“essential” 248. Of note, they only utilized an insulin 2 transgene. Insulin 2 differs
from insulin 1 for two amino acids (as well as multiple additional
polymorphisms in the leader and connecting peptide sequence). One of the amino
acids that differ between insulin 1 and insulin 2 is position 9 of the B:9-23
peptide (serine for insulin 2, proline for insulin 1), and we have in vivo
evidence that the immune response to the two peptides can be dramatically
different 275. A double insulin gene knockout NOD mouse (insulin 1
and insulin 2) with a mutant preproinsulin transgene to prevent metabolic
diabetes prevented diabetes4. The dramatic effect of insulin gene knockouts is consistent with
multiple studies indicating induction of widespread insulin gene expression
prevents diabetes, including expression in bone marrow-derived cells 276, 277.
Multiple
islet molecules are the target of autoantibodies or T cells in man and animal
models41, 240, 278, with the islet Zinc transporter (ZnNt8) a recent addition279.
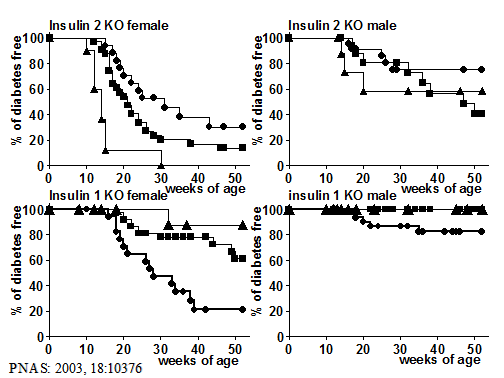
Figure 4. Life table
analysis of progression to diabetes of NOD mice with insulin 1 or insulin 2
gene knocked out, for male and female mice. Homozygous knockout mice are
represented by the triangles. Heterozygous knockout by the squares, and wild
type at the insulin gene by circles. Knocking out insulin 2 accelerates
diabetes whereas insulin 1 knockout prevents the majority of progression to
diabetes.
There is not universal
agreement if any of the target molecules are essential for immune mediated beta
cell destruction, though studies from the laboratories of Kay5, 280, Boitard104, 274, Jaeckel248 our group and others, strongly implicate immune responses to insulin as
a central component of type 1 diabetes of the NOD mouse. Our studies both in
man and mouse have concentrated on insulin30. Kay and coworkers utilizing a
transgene driving proinsulin expression with an I-E promoter completely prevent
diabetes and of interest abrogate development of IGRP CD8 reactive T cells, and
even prevent diabetes in a TCR transgenic targeting IGRP5, 280. Of note a similar I-E promoter
transgene but driving IGRP expression has no effect on progression to diabetes280, and thus they concluded that immune responses to proinsulin are
“upstream” of IGRP T cell targeting and crucial for diabetes, and lack of
immune response to insulin even prevented diabetes in mice with an anti-IGRP T
cell receptor transgene5.
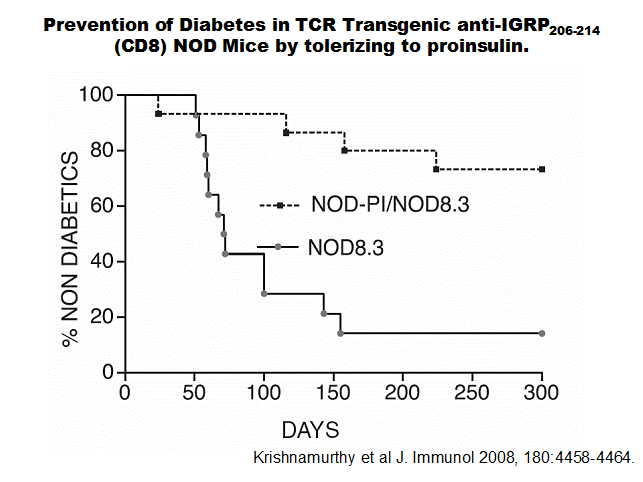
We have analyzed NOD mice
lacking both insulin 1 and 2 genes, with multiple transgenic founders with
either a native insulin sequence or a sequence with a single amino acid altered
(B16:A replacing B16:Y). These mice are
protected from diabetes and development of insulin autoantibodies and insulitis
are markedly decreased4. Restoring the native B:9-23
sequence with an islet transplant(but not bone marrow transplant) or peptide
immunization, or a native proinsulin transgene, restores anti-insulin
autoimmunity and generates CD4 T cells able to cause diabetes103.
BioBreeding Diabetes-Prone (BB-DP) Rat
The first inbred animal model for spontaneous, type 1
diabetes evolved from the discovery of diabetic animals in an outbred colony of
Wistar rats maintained at the Bio Breeding Research Laboratories in
Derivation of BB Rats
All BB rat colonies worldwide were derived from a
progenitor stock in
Genetics of BB Rats
It has long been known that diabetes of the BB rat
depends upon polymorphisms of the RT1U major histocompatibility
complex as well as a “peculiar” severe T cell lymphopenia inherited in an autosomal
recessive manner (lyp [lymphopenia] gene in BB rats that is unrelated to the
Lymphoid Tyrosine Phosphatase gene (LYP PTPN22) associated with human type 1
diabetes 155) 294, 295. Lymphopenia results from mutation of one of the Immune Associated
Nucleotide Related genes (IAN4). The
frameshift mutation of IAN4 is associated with increased apoptosis,
mitochondrial dysfunction296, and potentially heightened T cell receptor signaling297. In the mouse IAN family genes
are predominantly expressed in lymphocytes and expression is upregulated with
thymocyte differentiation298.

Figure
5. The lymphopenia gene (IDDM1 of rat) of BB-DP rats (Ian gene).
Essentially
all rat models of type 1 diabetes have the permissive RT1u MHC
haplotype 299(One exception reported in 1990 induced diabetes with thymectomy and
sublethal radiation of PVG/c stain rats 300). Linkage of diabetes to the MHC for BB rats was first defined by Colle
and coworkers in an analysis of diabetes incidence among F2 animals from a cross of BB-DP with Lewis strain rats
283. Lewis rats are not prone to diabetes and are
incompatible with the BB-DP at the MHC. All diabetic F2 animals were shown to be homozygous for the BB-DP
MHC which is the susceptible RT1u haplotype.
Multiple
BB rat disease-associated loci (iddmn)
have been reported 301-303. The lyp locus was mapped to a syntenic region of rat chromosome 4, is
designated iddm1, is responsible for the severe T cell loss when present in the
homozygous state 285, (identified as a frameshift
mutation of the IAN4 gene 304, 305).
Although
the contributions of the iddm1 (lyp) and idddm2 (MHC) loci to disease are
quantitatively large, they are not, in and of themselves, sufficient for
disease. Fixing the RT1u and
lyp alleles when breeding the BB-DP rat to diabetes-resistant ACI 294 and PVG 306 strains did not result in diabetes. Therefore, other loci are clearly
involved in rendering the animal susceptible to autoimmunity. Reports link
iddm4 (now termed iddm14307) with diabetes susceptibility in crosses between disease-resistant
Wistar-Furth (WF) and BB-DR animals which do not normally develop spontaneous
disease 301. The BB-DR animals are genetically similar to the BB-DP rat yet are
resistant to spontaneous type 1 diabetes due to the lack of the iddm1 locus(lymphopenia
gene). Thus the BB-DR animals have normal peripheral T cell numbers. However,
autoimmune-susceptibility genes are clearly present in the BB-DR as type 1
diabetes can be induced in BB-DR animals with immune manipulations if
administered in a short time window of 25-35 days of age. These manipulations
include viral infections 308, poly IC injections 309 and depletion of RT6+ T cells along with
an environmental trigger 310 and thymectomy311. One of the most fascinating
BB-DR related models is the induction of diabetes with the Kilham rat virus293, 308, 312-314. The ability of this virus to
induce diabetes in BB-DR rats was discovered following the spontaneous
infection of BB-DR colony with the virus.
The virus may act without infection of islet beta cells, with evidence
that induction of disease follows TLR9 mediated induction of a series of
cytokines and in particular IL12p40.
Chloroquine therapy decreases the development of diabetes293. Genetic loci influencing induced diabetes of the BB-DR and related
strains have been defined303, 315 with TCR beta(V beta 13) as one candidate gene for IDDM4307, 316. Backcrosses of (WFxDR-BB)F1 to WF rats resulted in approximately one-half of the
pups susceptible to type 1 diabetes induction. Furthermore, this susceptibility
mapped strongly to the iddm4 locus on chromosome 4 301. This 2.8-cM region on rat Chromosome (Chr) 4 locus contains several
major autoimmunity loci including aia2, aia3, and cia3, and it has been
assigned to a 2.8cM region proximal to Lyp/Ian4l1 317. These data,
as well as studies in the BB/OK strain 302, 318, crosses between BB-DP and non-autoimmune-prone rats 319, and RT1u
congenic strains 299 support the contribution of non-MHC loci in general susceptibility to
autoimmunity. Two other loci found to be associated with type 1 diabetes in the
BB-DP rat, include the diabetes-susceptible iddm3 and diabetes-resistant iddm5
loci 302, 318.
IDDM14
is localized to approximately 2 million bases on chromosome 4 containing the T
cell Receptor beta V-gene locus. This
diabtogenic locus is critical for development of diabetes not only for the BBDP
and BBDR strains but also for the LEW.1R1 strain where similar to BB-DR rats
diabetes is not spontaneous but can induced with agents such as poly-IC. Resistant strains at IDDM14 are WF, BN, and
F344. Two additional strains that
develop autoimmune diabetes (PVG.R8 and KDP) have not formally been evaluated
for linkage to IDDM14, but share a region defined by single nucleotide
polymorphisms with the susceptible strains, including mutations in several
Tcrb-V genes, (e.g. Tcrb-V13)320. This leads to the hypothesis,
not yet proven, that development of diabetes in these strains may be critically
dependent upon a specific T cell
receptor Vb sequence. This would be analogous to the hypothesis
that in NOD mice the Valpha sequence TRAV5-D4 associated with T cell receptors
targeting insulin peptide B:9-23 may be critical for insulin autoimmunity and
diabetes (though in this case the locus is not polymorphic between strains)107.
Immune Dysfunction in the BB-DP Rat and Its
Relationship to Lymphopenia
Lymphopenia was
first defined in 1981 by Jackson and colleagues 321. An early important characteristic distinguishing
lymphopenic from nonlymphopenic animals was the absence of the RT6+ subset of
peripheral T cells 322, 323. The under-representation of the CD45R+ T
cell subset isoform was also documented 324. The absence of RT6+CD45R+ T
cells correlated well with the overall reduction in the numbers of peripheral T
cells found in the BB-DP. Since thymocytes are both RT6- 322 and 98% CD45R- 325, a plausible role of the lymphopenia gene is to retard T cell
maturation at a stage prior to the expression of these peripheral T cell
antigens. The RT6+ population is known to contain regulatory
function since 1) depletion of the RT6+ population along with poly
I:C injection induces type 1 diabetes in BB-DR rats 326, 2) diabetes can be induced by adoptive transfer of
BB-DR lymphocytes into athymic nude WAG rats only if the BB-DR lymphocytes are
pretreated with anti-RT6 antibodies while cotransfer of RT6+ T cells prevents
diabetes 327 and 3) injection of RT6+ cells from BB-DR rats into BB-DP
lymphopenic rats prevents their spontaneous development of type 1 diabetes 328. Both CD4+CD25+ and CD4 T cells that express neither
CD25 nor Foxp3 have regulatory function329, 330.
Studies of the
immune function of lyp T cells from BB-DP rats report contradictory results of
poor in vitro responses and hyper-activated phenotypes. Early studies
report weak proliferative 331 and cytotoxic responses 287 to alloantigen as well as altered CD4-mediated signaling 332. Given the reduced numbers of T cells in the lyp
animals, especially in the CD8 compartment, the differences in responses in
these bulk read-out in vitro assays may be due to differences in
functional T cells present. It is now known that many of the peripheral T cells
in the BB-DP are undergoing apoptosis and therefore may not be functional,
although present, in these assays. More recent studies suggest that lyp T cells
show increased signs of activation ex vivo, including increased
expression of CD25 333 and OX40 activation markers 334, increased INFg
production 335, and increased mRNA of T cell signaling adapter
protein vav 336. The lyp cells also show increased signs of spontaneous proliferation ex
vivo with 1) 2X number of cycling cells as determined by increased DNA
content 337, 2) a 90% reduction in levels of cell-cycle inhibitor p27kip, increased levels of PCNA and phosphorylated Rb 338, and 3) incorporation of BrdU into >90% of lyp T cells compared to
~30% of normal T cells in a 13-day period 339. Collectively these data suggest that lyp T cells go through DNA
synthesis prior to their imminent demise although no studies have investigated
progression through mitosis or cell division. The resultant loss of T cells, as
opposed to an accumulation, suggests this proliferative state is
non-productive.
In addition to T
cell abnormalities associated with the lyp gene, studies of non-T cell subsets
of the BB-DP immune system suggest thymic and APC defects similar to the NOD
mouse model. A lower number of splenic DCs with decreased expression of MHC
class II and costimulatory ligand CD80 340 as well as decreased clustering
which improves stimulating capacity of DCs is reported 341. In addition, defects in the NK cell population 342 as well as thymic B cells 343 have been observed. The data support an overwhelming defect in
peripheral T cell regulation as a mechanism for disease and reports of enhanced
proliferative responses to superantigens following in vivo
administration 288 support the conclusion of defects in peripheral
tolerance in lyp rats. Few studies on thymic tolerance have been reported in
the BB-DP model although alterations in thymic medullary 285 and cortical 344 architecture have been associated with thymic tolerance defects were
noted.
A remarkable phenotype
afforded by the lyp mutation is the severely shortened lifespan of the lyp T
cells. Although thymic development appears normal in lyp animals with an almost
normal distribution of double-positive and CD4+ and CD8+
single-positive subsets which vary depending on background strain 306, peripheral T cell numbers are drastically reduced. The CD8+
T cell subset is nearly absent in the spleen and lymph nodes of lyp animals
with a few cells expressing lower levels of CD8 345, suggesting a faster death following thymic
selection than CD4+ T cells 346, 347. Tracking the export of T cells from the thymus with
fluorescent-labeling, Zadeh and coworkers measured the lifespan of CD4+ lyp T
cells to be less than one week 337. In addition, only few labeled thymocytes expressed RT6, which is
normally upregulated 1-2 weeks following thymic export. The majority of the
labeled thymocytes expressed the Thy1 Antigen which is characteristic of recent
thymic emigrants (RTEs) in the rat 337. The thymus of the lyp rat also showed reduced thymic export of RTEs,
data which is consistent with increased death of thymocytes in adult thymic
organ cultures (ATOCs) from lyp rats 348. Further data supporting an overwhelmingly shortened lifespan of lyp T
cells is the rapid loss of T cells from lymph nodes (LNs) and spleen following
thymectomy in lyp rats compared to a fairly stable T cell pool in thymectomized
normal rats 349, 350.
The efficient
clearance of apoptotic cells by phagocytosis has made detection of apoptosis
difficult in vivo. Death by apoptosis in lyp T cells is rapid in
vitro 306, 339 with a majority (~80%) of the cells dying in overnight cultures
compared to negligible death (~20%) observed in cultured T cells from
nonlymphopenic rats. Advances in detection of apoptotic cells allowed
identification of these dying cells in vivo through TUNEL and
FITC-Annexin V staining assays 351. Further characterization identified the clearance of the apoptotic T
cells in lyp rats in the liver 342. In mice, dying apoptotic CD8+ T cells are cleared in the liver whereas
CD4+ T cells remain in the LN even when apoptotic 352. The dying cells were difficult to define as T cells due to their
detectable but low expression of TCR, CD8, CD4, B220 and HSA proteins on their
surface 342.
The vast reduction in
RT6+ cells along with a preponderance of Thy1+ cells suggests lyp impacts T
cell development at the RTE (Recent Thymic Emigrant) stage. The effects of the
lyp mutation appear to be cell autonomous and restricted to bone marrow cells
as demonstrated through lyp à normal bone marrow chimeras 353. In addition, transplants of normal thymus into lyp recipients do not
reverse the lymphopenia, pointing to the developing T cells themselves
harboring the genetic disturbance 354. It is noteworthy that the T cell loss, activation,
and death phenotype observed in the BB-DP rat is also observed in lyp congenic
rats from diabetes-resistant strains (Fisher-lyp and PVG-lyp) which show T cell
lymphopenia but not diabetes 306, 338.
The
reduced number and early death of the T cells and their necessary role in type
1 diabetes induction present a paradox in the BB-DP model. Although a lack of
regulation in the BB-DP rat is well-documented, less is known about the
properties of the effector T cells responsible for the beta cell destruction.
There is disagreement whether the short-lived RTEs are responsible for disease,
as suggested by disease prevention with adult thymectomy 350. However, the thymectomy is required during a distinct time period for
protection – when performed at 8 weeks of age animals still develop diabetes
even though the vast majority of RTEs) still undergo rapid death. Combined with
newer studies showing antigen activation can rescue lyp T cells from death both
in vivo 339 and in vitro 306, another model suggests that higher affinity autoreactive T cells are
activated by autoantigen, survive, and serve the effector functions necessary
for immune clearance. Isolation and characterization of the effector T cells
responsible for beta cell destruction are still necessary to differentiate among
these models.
Disease Prevention in the BB-DP Rat
As in the NOD mouse
model, numerous treatments have been shown to prevent diabetes in the BB-DP rat
model. Early immunosuppression to prevent effector T cell function, including T
cell depletion with antibodies 355-357, thymectomy 349, 350, 358 or FK506 359, 360 prevents diabetes in the BB-DP rat. Similar to the effects in the NOD
model, complete Freund’s adjuvant 361 and viral infections 362, 363 prevent disease in the BB-DP model as well. Reversing the defect in the
regulatory population also prevents diabetes. This reversal has been shown to
be successful in disease prevention with the addition of RT6+ cells 364-367 or with human TNFalpha, a cytokine which influences regulatory
properties of T cells 368 . Although alterations in peripheral tolerance are well-documented in
the BB-DP, little is known about alterations in thymic tolerance in this model.
However a report that in thymic organ culture of BB-DP thymocytes, coculture
with islets, but not thyrocytes, prevents diabetes and reduces insulitis upon
adoptive transfer 369. These data suggest diabetes can be reversed with increased thymic
tolerance. The non-MHC and non-lyp loci may indeed influence the repertoire of
autoreactive T cells and distinguish lyp animals susceptible or resistant to
autoimmunity.
BB rats raised in a
germ-free environment develop diabetes.
Nevertheless recent studies indicate that treatment of diabetes-prone BB
rats with antibiotics which change their GI flora (with correlation with
diabetes outcome), especially when combined with a casein free diet are
protected from the development of diabetes370.
Long Evans Tokushima Lean (LETL) Rat Model (Komeda
Diabetes-prone)
An additional rat
model of spontaneous development of type 1 diabetes was described in 1991 371. Inbreeding of a rat showing signs of diabetes in
1983 resulted in the LETL strain maintained at the Tokushima Research
Institute. These rats have the advantage of spontaneous disease incidence
without a gender bias or a requirement for lymphopenia. Like the NOD mouse,
infiltration of the pancreas, salivary, and lachrymal glands is observed 371. The disease shows a polygenic mode of inheritance372 including a large contribution of the MHC RT-1u haplotype shared with the BB-DP rat 371. Selection of the Komeda Diabetes-Prone rat
substrain improved disease penetrance, with 100% insulitis and 70% diabetes by
120 days of age 373. A genome-wide scan of this strain identified a
novel non-MHC associated disease locus on rat chromosome 11 which was shown to
be essential for development of insulitis 373. Mutated Cblb
alleles when introduced on a non-KDP RT1(u) rat resulted in a low incidence of
diabetes and thyroidits, suggesting additional modifier genes influence
penetrance374. An additional Komeda
Non-Diabetic (KND) substrain was established which, although genetically very
similar to the KDP strain, does not develop type 1 diabetes 375. This strain serves as an important control in both genetic and
immunological studies of the KDP rat. This rat model thus most closely
resembles human type 1 diabetes in its spontaneous onset, lack of gender bias,
lack of lymphopenia, and association with MHC class II. The non-MHC mutation
segregating as a recessive is a nonsense mutation of the Cblb gene (Casitas
B-Lineage Lymphoma b gene) which is important for T cell regulation 376. Cbl-b is a ubiquitin ligase important for CD28 co-stimulation during T
cell activation. Analysis by two groups for polymorphisms of the human Cbl-b
gene failed to find an association with human type 1 diabetes 377, 378. It does not appear that Cbl-b
locus contributes to development of type 1 diabetes of man though there is one
report of an interaction with CTLA-4377-379.

Figure 6. The Cblb mutation of the Komeda diabetic rat.
LEW.1AR1/Ztm- iddm rat
In 2001 Lenzen and coworkers
described a new rat model of insulin deficient diabetes in which approximately
20% of the animals (no sex bias) developed diabetes characterized by lymphocytic
infiltration of the pancreas from rats that were not lymphopenic 380, 381.
Similar to other rat strains with immune-mediated diabetes, these animals had
RT1 haplotype with u alleles (RT1.Aa B/Du Cu 380 and is
a recombinant RT1 haplotype (RT1r2). In addition to islets, larger
pancreatic ducts show mononuclear infiltrates. The islet infiltrates are
predominantly CD8 T cells (>50%) with a relatively small percentage of CD4
cells (4%), and many ED1-positive macrophages 381. Transfer
of CD8+ T cells from prediabetic mice (but not CD4+ cells) protects 382. At early stages of infiltration macrophages
predominate383. Of note non-diabetic rats lacked islet
infiltration, with none of the non-diabetic animals having infiltrates! Other
organs, including endocrine organs did not have infiltrates. In summary, the
LEW1.AR1/Ztm-iddm rat is a novel model of islet destruction in the presence of
islet infiltrates that differs from other models in that age-matched animals
lacking diabetes did not have infiltrates. This suggests an acute disease
process and one with dramatic heterogeneity between rats.
TCR Tg (transgenic)
and retrogenic Mouse Models of Type 1 Diabetes
Early studies
breeding non-pancreatic specific TCR transgenics, either class-II restricted
(D011.10 anti-OVA) or class I-restricted (2C anti-Ld) onto the NOD background resulted in no change in
disease incidence 384. Although the transgenes showed good allelic
exclusion at the beta locus, the alpha locus was leakier allowing endogenous
receptor expression. This finding supported selection for pancreatic-specific T
cells in mice skewed T cell repertoires with innocuous receptors.
A number of T cell
clones have been isolated from the spleens of diabetic NOD mice (BDC2.5,
BDC6.9) 385, and the pancreata of pre-diabetic 238, 386-388 NOD or RIP-B7.1 NOD transgenic 389 mice and islet-transplanted diabetic 390 NOD mice, and from islets of NOD mice (BDC12-4.1) 238. Both CD4 and CD8 clones have been generated 391 which have varying abilities to induce or suppress insulitis and
diabetes 392. The TCR (T Cell Receptor) of many of these clones
have been utilized to produce TCR transgenic (Tg) mice on various backgrounds264. A summary of TCR transgenic diabetic mice is listed in Table 3.4. A major advance has been the development of
the technology to produce retrogenics mice107, 264, 393-397 that has rapidly been applied to diabetogenic T cell receptors.
Table 3.4 T Cell Receptor transgenic NOD Mice
|
T Cell Receptor transgenic |
Vbeta |
Valpha |
CD4/CD8 |
Antigen |
Disease Phenotype |
|
D011.10 |
8.2 |
Endogenous |
CD4 |
OVA/IAd |
No
change in diabetes frequency |
|
2C |
8.2 |
3.1 |
CD8 |
Ld |
No
change in diabetes frequency |
|
BDC2.5 |
4 |
|
CD4 |
Chromagranin |
Rapid,
uniform insulitis at 3 weeks of age; reduced diabetes incidence; higher
incidence of diabetes with CD1d deficiency (?NK T cell protection) and in
BDC2.5 C57Bl.IAg7 congenic mice which maps to Idd7 resistance in NOD
mice; accelerated diabetes on NOD SCID background |
|
BDC6.9 |
|
|
CD4 |
Unknown
AutoAg on chr 6 in NOD and SWR pancreas |
Accelerated
diabetes on NOD background positively
selected on I-Ag7 |
|
BDC12-4.1 |
2.1 |
13.3 (TRAV 5D-4) |
CD4 |
Insulin
B:13-23 |
Positively
selected on I-Ag7 but animals lymphopenic; Diabetes
observed on Rag-/- background; Insulitis on Mixed
strain Rag+ background. |
|
4.1-NOD |
|
|
CD4 |
Unknown
AutoAg in pancreas |
Positively
selected with IAg7; negatively selected with other
class II molecules; Kill via Fas. Increased diabetes incidence; same
frequency Rag-/- background - CD8
independent. |
|
8.3-TCRb |
8.1 |
Endogenous |
CD8 |
IGRP-beta
cell specific |
Accelerated
diabetes with reduced period of benign insulitis; NRP-V7 peptide tetramer for diabetes
prediction in NOD. |
|
8.3-NOD |
8.1 |
Va17/Ja42 |
CD8 |
IGRP |
Accelerated
diabetes with reduced period of benign
insulitis; Kill by Fas exclusively. On rag-/- background get decreased
disease incidence - requires CD4 help. |
|
9.33 |
6 |
19 |
CD8 |
Unknown
AutoAg in pancreas |
Accelerated
diabetes and increased incidence; requires
CD4 help. No diabetes on scid background – not due
to lack of CD4 but rather lack of escape from negative
selection with only one TCR |
|
NOD.AI4 |
2 |
8 |
CD8 |
?DMK |
DMK
widely distributed molecule, mimotope evidence re: target; Needs both Kd and Db
for killing; accelerated diabetes in absence of CD4 help or B7.1 expression. |
|
GAD206;GAD524 “Retrogenic” |
|
|
CD4 |
GAD524-543
Subset |
Vignali
GAD reactive TCR “retrogenic” NOD mice with retroviral stem cell gene
transfer. No diabetes TCR’s induced encephalitis. |
|
|
|
|
|
|
|
The BDC2.5 TCR
transgenic mouse was generated from the BDC2.5 CD4 T cell clone (Ag) 398 which Haskins and coworkers have identified targets
the WE-14 peptide of chromogranin clones BDC 10.1 and BDC 5.10.3 also targets
chromagranin 2. The BDC2.5 TCR transgenic mouse develops a uniform insulitis at 3
weeks of age followed by a low incidence of overt diabetes. Surprisingly,
BDC2.5 transgenic mice on the C57BL/6.IAg7 background showed a higher incidence of diabetes
than on the NOD background. The difference in disease incidence was mapped to
the Idd7 disease-resistance interval in the NOD 66. The BDC2.5 TCR transgene bred onto the NOD.scid
background resulted in a much accelerated onset of type 1 diabetes, suggesting
regulatory cells prevent diabetes induction in the normal NOD setting. Of note,
diabetes is restored in the absence of NKT cells with deletion of the CD1 locus
399. BDC2.5 T cells have been
utilized to create potent regulatory T cells that are able to suppress the
spontaneous disease of the NOD mouse213, 400.
Another CD4+ clone, BDC6.9, isolated in Kathryn Haskin’s
laboratory has been made into a TCR transgenic mouse. Unlike the BDC2.5
transgenic mice, BDC6.9 transgenic mice develop early diabetes by the 3rd
and 4th generation backcross to the NOD background 178. The advantage to this clone is the ability to
manipulate the presence and absence of self-Ag, as the stimulatory Ag is found
in pancreas from NOD and SWR mice, but not other common strains of mice.
Daniel and Wegmann
isolated a series of T cell clones from islet infiltrates and found that the
majority reacted with insulin and the great majority of insulin-reactive
(>90%) CD4 clones reacted with the insulin peptide B:9-23 (B chain, amino
acids 9-23). A transgenic T cell receptor mouse has been established with the
12-4.1 T cell receptor anti-B:9-23 clone. This T cell recognizes insulin
peptide sequences 13-23, using the common alpha chain motif (Valpha 13.3
(TRAV5D-4*04) with Jalpha 53)401. As a retrogenic the T cell receptor of the 12-4.4 clone is also able
to induce insulin autoantibodies and diabetes 107, 397. The 12-4.1 and 12-4.4 alpha
chains have the same TRAV5D-4*04 T cell receptor segment, related J alpha
segments, but different alpha chain N region and different Vbeta segments. The ability of multiple T cell receptors with
the related alpha chains that target the insulin B:9-23 peptide to induce
insulin autoantibodies and diabetes has led to the hypothesis that such
targeting is primarily driven by the shared alpha chain germline sequences 32.
Both Class II-restricted CD4+ and class-I restricted CD8+ T cell clones isolated in the laboratory of Santamaria have been developed into TCR
transgenic mice with either beta chains alone or in combination with alpha
chains. A majority of CD8+ T cell
clones isolated from the pancreas of pre-diabetic NOD mice show the same TCR Va17/Ja42 with N region sequence M-R-D/E, which has been
used to generate 8.3 TCRab transgenic mice. The Vb8.1 CD8+ TCR transgenic mice, transgenic for the TCR
beta chain only, and 8.3 CD8+ transgenic mice show an accelerated incidence of
disease with a reduced period of benign insulitis 386. In the 8.1 beta-chain only transgenic mice, the
islet-reactive T cells were found to have the same alpha chain as the original
clone, suggesting a selection of this clone in diabetogenesis 386. The target antigen of the 8.3 clone is the molecule
IGRP (islet-specific glucose-6-phosphatase catalytic subunit-related protein)
and an excellent tetramer reagent (using a mimotope of the native peptide
NRP-V7) allows staining of reactive T cells from peripheral blood of NOD mice,
with numbers of T cells correlating with disease progression 208. Islet-reactive CD8+ T cells increase their avidity binding to the 8.3-reactive
peptide presented in Kd as
shown through tetramer analysis 209. Kay and coworkers have recently
found that the 8.3 transgenic induction of diabetes is dependent upon an immune
response to insulin by blocking such a response with a proinsulin transgene5.
The NY4.1 transgenic mouse is from a CD4 clone. This
TCR transgenic mouse has the same disease incidence on RAG2 KO or normal NOD
background, confirming the ability of some CD4+ cells to induce diabetes in the absence of CD8+ T cells 386. However, breeding the Vb8.3 CD8+ TCRabtransgenic mice onto the RAG2 KO background
results in greatly reduced disease onset and demonstrates a role for CD4 cells
in the efficient recruitment of CD8+ T cells to islets 387. The NY4.1 as a retrogenic induces diabetes264. The transition from benign insulitis to overt diabetes appears to be a
loss in the ability to regulate autoreactive cells. Epitope spreading and
affinity maturation have been shown to be important in this process.
Previous reports demonstrated that a CD8+ T cell clone (G9C8) isolated from the pancreas of
RIP-B7.1 transgenic mice did not require CD4 cells for disease transfer 389. In a more recent study a TCR transgenic mouse
(NOD.AI4) was generated from a CD8 clone which was isolated from an
unmanipulated NOD pancreas. The transgenic T cells were capable of inducing
diabetes free of CD4 help or transgenic costimulation, supporting the idea that
high-affinity CD8 T cells are independently capable of beta cell destruction 388. Of interest the clone apparently requires both Kd and Db to be present
for cytotoxicity 84. However, another CD8+ islet-reactive TCR transgenic, the 9.3 transgenic,
showed accelerated diabetes compared to normal NOD mice but no diabetes on the
NOD.scid background 402. The unusual finding in this transgenic system was the necessity for
two dissimilar receptors on the cell surface for disease incidence as
identified by co-staining with antibodies specific for the transgene and a mix
of other Vbeta-specific Antibodies. The authors conclude that the lowered
expression of the pancreatic-specific receptor due to coexpression of other
TCRs allowed escape of the transgenic receptors from negative selection.
Adoptive transfer studies showed these cells did require CD4 help for diabetes
induction 402.
Vignali and coworkers have
introduced a powerful new technique for the study of T cell receptor expression
that they term “retrogenics” in distinction to transgenics. The use retroviral-
mediated stem cell gene transfer of T cell receptor sequences and bone marrow
transplantation of the transduced bone marrow. For the production of retrogenic
mice a retrovirus is used to introduce genes coding for T Cell receptor chains
into bone marrow cells that are then transplanted into immunodeficient mice
(usually SCID or rag-/-). Within weeks
one can observe the development of insulin autoantibodies, insulitis and
diabetes. The figure (Nakayama) below
illustrates the general methodology for production of transgenic mice where one
can introduce a series of Valpha chains of anti-islet T cell receptors that differ in different segments or complete
alpha a beta chain T cell receptors. If the donor marrow is from a Calpha
knockout mouse, the only alpha chain produced in the retrogenic mouse will be
the introduced chain. If the donor mouse
is a SCID and complete alpha and beta TCR chains are introduced, only a single
T cell receptor will be produced.
Vignali and
coworkers studied two T cell receptor positive controls and showed that BDC2.5
and 4.1 retrogenic bone marrow induced diabetes in NOD-scid mice. In contrast T
cell receptors for GAD206-220 and GAD524-538, though reactive cells were
present in vivo, failed to influence the development of diabetes of NOD
mice 396. A subset of anti-GAD TCR
retrogenic develop encephalitis (no insulitis).
More generally the retrogenic technique provides a powerful tool to
rapidly study many T cell receptors in vivo264 . Studies of multiple T cell
receptors targeting different islet autoantigens indicate that only a subset
lead to insulitis and less produce diabetes.
T cell receptors targeting the insulin B:9-23 peptide can cause disease,
and a set of T cell receptors are more diabetogenic that react with currently
unknown islet autoantigens. It is likely
that production of retrogenics will accelerate the study of T cell receptors
and importantly segments of T cell receptors to determine targeting and
pathogenicity. For instance DiLorenzo
and coworkers have utilized retrogenics to produce large numbers of CD8 T cells
with specific T cell receptors to study in vitro393. A disadvantage of the retrogenic technique is that the introduced T
cell receptors are naturally not heritable, and for each experiment new mice
need to be created.
Transgenic Expression NeoSelf-Antigens in Pancreas
A limitation for many of the studies
using TCRs from pancreatic-reactive clones is the lack of knowledge of the
antigen. This is a limitation that is rapidly being addressed, now with
anti-insulin TCR transgenics (BDC12-4.1 TCR), IGRP (islet-specific
glucose-6-phosphatase catalytic subunit-related protein) transgenics (8.3 TCR
transgenics) 207, AI4 transgenics (?DMK target autoantigen) and BDC
2.5 and BDC10.1 transgenic (chromogranin) 244. Nevertheless, studies with defined antigens introduced into the
pancreas by transgenesis on the RIP (rat insulin promoter) promoter, combined
with T cell receptor transgenics have provided an elegant system for evaluating
tolerance to islet expressed molecules. A list of models is present in Table
3.5. A important caveat, not recognized in the very initial studies of Rat
Insulin Promoter driven Neo-Antigen expression is the expression of the
transgene neoSelf-Antigen in lymphoid tissues such as the thymus. Thus
autoimmunity can be influenced not only by antigen in the target tissue but
dramatically by central tolerance mechanisms. The initial studies by Miller and
coworkers with RIP induced expression of a “foreign” class I molecule,
elegantly demonstrate the importance of minute expression within the thymus of
neo-Self-Antigen, with tolerance dependent upon thymic expression of the
antigen and not expression within islets 403.Transgenic expression of viral LCMV under the RIP promoter is
associated with ignorance of the T cells to the LCMV antigen unless prompted by
LCMV systemic infection which leads to diabetes 404, 405. It is hypothesized that this ignorance is due to poor
cross-presentation of antigens such as gp33 by CD4 T cells 406. The diabetes in this model always requires CD8+ T cells whereas CD4+ T cells are only required in some lines 407-409. When the RIP-LCMV is crossed with a TCR transgenic mouse specific for
LCMV, the animal still requires systemic LCMV infection for diabetes induction,
although in this model the disease onset is accelerated compared to the non TCR
transgenic model 405. In the LCMV transgenic model, the CD8+ T cells do not require CD4+ help as suggested by disease incidence with LCMV
infection in class II-deficient 408 and CD40L-deficient animals (120). As with other systems, crossing the RIP-LCMV with
RIP-B7.1 transgenic mice, which provides costimulation directly on islet cells,
results in spontaneous diabetes 410, 411. Recent work with a defined LCMV protein (NP) crossed with a RIP-IL4
transgenic mouse showed protection from diabetes following LCMV-infection 412. The IL4 was shown to inhibit the acquisition of cytolytic activity yet
promote expansion and survival of CD8+ T cells through an increased expression of B7.2 on
DCs 412.
Another
neo self-Antigen model targets expression of flu hemagglutinin (HA) antigen to
pancreatic beta cells using the RIP promoter. In one set of models HA expressed
with Igk promoter results in
expression in thymic medullary and cortical epithelial cells (as wells a B
lymphocytes)
and thymuses induce CD25+
regulatory T cells 413. Unlike the LCMV protein, the Ins-HA mice appear tolerant to the HA
Antigen following flu immunization if administered in adulthood 410, 414. A CD8+ TCR
transgenic specific for HA (CL4) can adoptively transfer diabetes in Ins-HA
recipients if the transgenic T cells are pre-activated in vitro 415. In addition, CL4xIns-HA double transgenic mice are not tolerant and
get spontaneous diabetes by 8 days of age with few T cells in the islet
infiltrate 414. Further work on the CL4 TCR showed this TCR
possessed an unusually high affinity to HA/Kd.
Table
3.5 “Neo-Self Antigen Models to Type 1 Diabetes
 This high affinity is evident
in the activation of the TCR in the absence of costimulation or with soluble
monomeric complexes 415. Like this CD8+ TCR
transgenic mouse, tolerance was also not established to the HA Antigen in mice
doubly transgenic for the Ins-HA transgenic and a CD4+ anti-HA TCR transgenic 416. Other HA-specific CD4+ TCR transgenic mice crossed with Ins-HA developed
varying degrees of diabetes depending on the background strain 416, 417. Recently, the Ins-HA model has been used in studies of peripheral
tolerance of CD8+ T cells
in various mouse studies. Defects in peripheral tolerance in NOD mice compared
to BALB/c and B10.D2 were confirmed by showing higher avidity binding of NOD T
cells to Kd-HA tetramers 205.
This high affinity is evident
in the activation of the TCR in the absence of costimulation or with soluble
monomeric complexes 415. Like this CD8+ TCR
transgenic mouse, tolerance was also not established to the HA Antigen in mice
doubly transgenic for the Ins-HA transgenic and a CD4+ anti-HA TCR transgenic 416. Other HA-specific CD4+ TCR transgenic mice crossed with Ins-HA developed
varying degrees of diabetes depending on the background strain 416, 417. Recently, the Ins-HA model has been used in studies of peripheral
tolerance of CD8+ T cells
in various mouse studies. Defects in peripheral tolerance in NOD mice compared
to BALB/c and B10.D2 were confirmed by showing higher avidity binding of NOD T
cells to Kd-HA tetramers 205.
Similar
to the Ins-HA model, RIP-OVA transgenic mice can become diabetic when
transferred with pre-activated anti-OVA CD8+ T cells 418. In addition, early diabetes is observed in OT-1 (TCR transgenic
anti-OVA)xRIP-OVA double transgenic mice, suggesting a lack of tolerance and
ignorance in this model 419. Another model places the class I Kb molecule under control of the RIP-promoter 403. Crossing the RIP-Kb mice with a CD8+ TCR anti-Kb transgenic mouse shows tolerance to this pancreatic
islet, which is associated with both thymic deletion of high-affinity clones by
residual thymic expression and functional unresponsiveness of remaining clones.
This tolerance can be broken, resulting in diabetes induction, with RIP
expression of IL2, a cytokine which has been shown to reverse T cell anergy 420.
Transgenic expression of the exogenous hen egg lysozyme
(HEL) protein by the RIP promoter, together with an anti-HEL TCR transgenic,
shows thymic deletion and peripheral anergy of transgenic cells on the C57BL/6
background. However when bred onto the NOD background, maintaining the
necessary H-2k MHC, the HEL
model showed defects in both thymic deletion and T cell anergy induction
leading to overt diabetes in the NOD RIP-HEL/TCR double-transgenic mice. This
model clearly demonstrates the influence of NOD non-MHC genes on tolerance
processes 421.
As mentioned above, transgenic mice expressing the
costimulatory molecule B7.1 on pancreatic islet cells (RIP-B7.1) have been
useful in studying “spontaneous” diabetes by crossing this mouse with models
which otherwise do not show disease incidence. This mouse on the C57BL/6
background does not get diabetes but can become diabetic when crossed with
RIP-LCMV, RIP-TNFa,
or RIP-IE or with mice expressing human HLA molecules associated with type 1
diabetes. The usefulness of the RIP-B7.1 model is to address the sensitivity of
disease induction allowing the systems to be stressed to determine if diabetes
can occur. RIP-B7.1 transgenic expression on the NOD background results in
diabetes incidence as early as the first backcross although IAg7 homozygosity is required 415. The largest effect of
B7.1 expression is on the CD8 cells, thereby negating the necessity of CD4+ help although the presence of CD4+ cells accelerates the disease progression by aiding
in migration of cells to the pancreas. Unlike spontaneous NOD diabetes, B cells
are not required for disease in the RIP-B7.1 model 422.
Immunization/Immunoregulatory
Induced Models of Diabetes
For many experimental autoimmune disorders it is possible
to induce pathology by immunizing with the presumed target autoantigen,
creating disorders such as Experimental Autoimmune Encephalitis (EAE) 122. Until recently it has not been possible to induce immune mediated
diabetes in mice or rats with specific islet autoantigens. Immunization with a
heat shock protein was associated with transient hyperglycemia 423 and this molecule, despite its ubiquitous expression
is reported to be both an autoantigen and activator of the innate immune system
245.
There
is actually an “old” literature where insulin immunization of cows and rabbits
induced “insulitis” and even diabetes in rabbits 424-429but with the “difficulty” of the bovine model, and apparently problems
in reproducibly inducing diabetes in the rabbit model, (as available insulin
became purer), this area of investigation was not pursued. It was certainly
recognized that most patients treated with insulin (again becoming less with
improved insulin purity) produced insulin antibodies and immunoreactivity to
insulin was studied in mouse models 430 and man 431. It is somewhat surprising that it was not possible
to induce diabetes with proinsulin/insulin/or insulin peptides. Recent studies
indicate that insulin and its peptides can induce anti-islet autoimmunity
(insulin autoantibodies and insulitis), but in normal mouse strains (e.g.,
BALB/c, C57BL/6) this is not sufficient to engender diabetes. In mice carrying
a transgene inducing B7.1 (costimulatory molecule) expression on islets,
diabetes is routinely induced following insulin/ insulin peptide B:9-23
immunization given the appropriate MHC alleles 232, 432-435.
Figure 7. Induction of
insulin autoantibodies following subcutaneous injection of insulin peptide
B:9-23 in incomplete Freund’s adjuvant.
Mice
with I-Ad (e.g., BALB/c mice) or I-Ag7 (e.g. NOD mice) respond
to subcutaneous immunization with insulin peptide B:9-23 with the rapid
production of insulin autoantibodies (Figure 7) 432-434. NOD mice respond in a similar fashion, but because they already
produce insulin autoantibodies, one observes increased levels rather than de
novo production of autoantibodies. In NOD mice the expression of insulin autoantibodies
persist for months, rather than being transient following immunization with
B:9-23 peptide. These are actual autoantibodies in that they can be blocked
with “mouse” insulin, and the antibodies react with intact insulin and cannot
be absorbed with the immunizing B:9-23 peptide. Of note the response is MHC
restricted as shown by analysis of congenic strains and no other peptide of
proinsulin to date has been found to induce the autoantibodies.
When
NOD mice are immunized with the insulin peptide B:9-23 they also produce
antibodies reacting with the peptide itself, that depending on the chronicity
of administration can induce anaphylaxis 436, 437. Though insulin autoantibodies are induced by insulin peptide B:9-23
immunization of BALB/c mice, insulitis was not observed. To produce insulitis,
immunization of BALB/c mice was combined with two weeks of administration of
subcutaneous poly-IC. Poly-IC is a toll 3 receptor activator and mimic of
double stranded RNA. It induces circulating levels of interferon alpha, and
acts through induction of interferon alpha to induce diabetes438. Despite the development of both insulin
autoantibodies and insulitis, the BALB/c mice do not progress to diabetes. In
mice expressing B7.1 in islet beta cells, diabetes can be induced with either
poly-IC or insulin B:9-23 peptide alone, as well as with both together 433. Thus tolerance to insulin can be readily broken,
and there is a dramatic restriction in terms of the peptides able to induce
disease. We suspect that normal BALB/c mice are already sensitized to the
insulin B:9-23 peptide prior to immunization with this peptide given the
rapidity of the induction of insulin autoantibodies reacting with a
conformational epitope of insulin. For the C57Bl6 mouse that does not respond
to insulin peptide B:9-23 (I-Ab) disease can be induced in B7.1
islet transgenic mice by immunization with intact insulin, presumably by
reactivity to a different epitope of insulin 435. Induction of cytokines can also
accelerate the development of diabetes in NOD mice depending upon the stage at
which for instance NOD mice with established insulitis when infected with
rotavirus439.
Co-stimulation
As mentioned previously, the expression of the B7.1
molecule on islets enhances the diabetogenicity of a series of stimuli that by
themselves are insufficient to induce diabetes. This includes insulin peptide
B:9-23 (BALB/c mice) 433, poly-IC (C57BL/6 mice) 440, LCMV-GP peptide 441, LCMV-GP gp33 and preproinsulin encoding plasmid 442, human DR4 transgene 443, human DQ8 transgene 443, transgenic islet expression of IL2 444, and transgene induced expression of TNF alpha on beta cells 445. It was hypothesized the B7-H1 would be a negative regulator of
autoimmunity, but transgene induced expression of B7-H1 on islets also induced
diabetes 446.
“Humanized” Mice
Another group of animal models being developed in the
past decade for the study of type 1 diabetes are the humanized mice. There are
two general approaches: 1. Introduction
of human genes into mice and 2. Transplantation of human stem cells into
immunodeficient mice19. Both approaches represent
important technical advances20, 447, 448.
The
class II HLA molecule DQB1 chain with serine, alanine or valine at P57 of the b chain, similar to the NOD MHC class II haplotype,
confers the greatest risk for type 1 diabetes in humans. The DQ8 (DQA1*0301
plus DQB1*0302 (alanine at P57)) haplotype has been expressed in transgenic
mice 449. The DQ8 transgenic mice do not develop diabetes even when backcrossed
to the NOD background. However, when these mice are crossed with the RIP-B7.1
transgenic mice, 80% of the mice develop insulitis and type 1 diabetes whereas
control human DQ6 (diabetes-resistant haplotype)xRIP-B7.1 double-transgenic
mice do not develop insulitis or diabetes. The DQ8xRIP-B7.1 transgenic mice
have been used to map approximately 10 different T cell epitopes in human
GAD65, one of which is also a dominant T cell epitope in NOD mice restricted to
IAg7 450, 451. Transgenic expression of the human disease-associated DR4 allele in
mice showed surprisingly that this allele, in combination with the highly
susceptible DQ8 allele, actually protected mice from disease. Whereas 25% of
DR4/RIP-B7 mice developed diabetes compared to >80% of DQ8/RIP-B7 mice, the
triple transgenic DR4/DQ8/RIP-B7 had only a 23% disease incidence, showing a
dominant protective effect for the DR4 allele (137). The diabetes incidence afforded by the
immunostimulatory RIP-B7.1 transgene enables studies of human autoimmunity
genes in mice with the important caveat that the pattern of disease can be very
unusual as found by Lipes and coworkers with pancreatitis depending on the
strain in the absence of murine class II genes 452. RIP-B7/DRB1*0404 HLA transgenic mice spontaneously develop T cells
reacting with GAD65 and Glial fibrillary acid protein (GFAP) and approximately
20% develop diabetes at a mean age of 40 weeks453. A human T cell receptor transgenic mouse (anti GAD65/67 (555-567)
develops insulitis on a human DR4 rag-/- background268.
The introduction of human DQ8 onto the NOD background has
resulted in a series of autoimmune disorders. In particular a disorder
resembling dermatitis herpetiformis is induced followed immunization with
gluten 454, a disorder resembling polychondritis following immunization with type
II collagen 455.
Marron and coworkers demonstrated that transgenic
expression of the common human class I allele, HLA-A2.1, accelerated the
development of diabetes in NOD mice. This effect was not simply a result of
expression of any human class I allele, as HLA-B27 (associated with ankylosing
spondylitis in man) if anything decreased development of diabetes 456. The A2.1 mouse islets have been used to study a
model of human lymphocytes in NOD-Rag1 (null)Prf1 (null) mice rejecting these
islets. Transplanting both into the spleen enhanced graft and lymphocyte
interaction. When lymphocytes came from A2.1 positive donors the grafts
survived, while from HA-A2 negative donors, the grafts were rejected 457. Humanized HLA-A2.1 mice
(further refined to eliminate murine class I antigens) have been utilized to
identify peptides recognized by CD8 T cells of potential relevance to human
type 1 diabetes447, 458, 459.
Human Stem Cell Transplantation
It is likely that many additional human molecules will be
introduced into murine strains to “humanize” the immune system. An approach to utilize transplantation/transfer
of human cells directly into mice allows the rapid introduction of multiple
genes and cell types20. These studies have been greatly
enhanced with the development of common gamma chain knockout mice on the
NOD-scid background460. This combines severe adaptive immunodeficiency with abnormalities of
NK T cells and allows engraftment of both peripheral blood cells and stem cells
and the study of alloreactivity with transplant rejection19. This model has been combined
with the
Conclusion
The multiple animal models of type 1A (immune mediated)
diabetes amply demonstrate that there are many pathways that can result in
immune mediated (autoimmune) destruction of islet beta cells, and that the
spontaneous animal models each represent a different pathway. One can
generalize with the following observations:
- T cell destruction of islet beta cells can be
entrained by the targeting of single islet molecular determinants by T
cells with a single T cell receptor (and obviously also with multiple
receptors). In general however, both CD4 and CD8 T cells collaborate in
causing beta cell destruction. It is likely that each T cell receptor
(e.g., clone or transgenic) will follow its own rules, depending on the
avidity and other qualities of the receptor), in terms of whether
ignorance, tolerance or beta cell destruction results.
- There is often T cell receptor biased chain
utilization of the T cells targeting a specific peptide autoantigen.
- In the spontaneous animal models multiple islet
antigens are the targets of autoimmunity.
- It is likely in some common models, such as the
NOD mouse, a single determinant may be essential for disease, such as the
insulin sequence B:9-23.
- The finding that a given antigen is the target
of autoreactive T lymphocytes, and even that T cells targeting that
antigen can transfer disease, does not mean that the targeted molecule is
important for pathogenesis of the spontaneous disorder, and the best
current test for importance of a target epitope, is gene knockout or
mutation. If disease occurs at the same rate in the absence of the
molecule, it cannot be essential, though may contribute to the disorder.
- Multiple genes synergistically contribute to
autoimmunity and the MHC is often essential in determining the organ
targeted.
- Both pathogenic T cells and regulatory T cells
(and other regulatory cells) modulate the development of disease.
The tremendous increase in knowledge relative to the
animal models will hopefully translate into informed trials for the prevention
of type 1A diabetes in man and long-term success of islet transplantation.
There is an important opportunity to attempt to develop assays and paradigms in
the animal models that are directly applicable to man. In particular in man one
must sample the peripheral blood for disease prediction, and every effort
should be made in the “simpler” animal models to develop assays of blood that
can be applied to man.
Reference List
(1)
Stadinski BD, Zhang L, Crawford F,
Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin
bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S
A 2010 Jun 15;107(24):10978-83.PM:20534455
(2)
Stadinski BD, Delong T, Reisdorph N,
Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in
type 1 diabetes. Nat Immunol 2010 Mar;11(3):225-31.PM:20139986
(3)
Lang J, Bellgrau D. Animal models of
type 1 diabetes: genetics and immunological function. Adv Exp Med Biol
2004;552:91-116.http://www.ncbi.nlm.nih.gov/pubmed/15622960?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(4)
Nakayama M, Abiru N, Moriyama H, Babaya
N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development
of type 1 diabetes in NOD mice. Nature 2005 May 12;435(7039):220-3.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=15889095
(5)
Krishnamurthy B, Mariana L, Gellert SA,
Colman PG, Harrison LC, Lew AM, et al. Autoimmunity to Both Proinsulin and IGRP
Is Required for Diabetes in Nonobese Diabetic 8.3 TCR Transgenic Mice. J
Immunol 2008 Apr 1;180(7):4458-64.http://www.ncbi.nlm.nih.gov/pubmed/18354167?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(6)
Su MA, Giang K, Zumer K, Jiang H, Oven
I, Rinn JL, et al. Mechanisms of an autoimmunity syndrome in mice caused by a
dominant mutation in Aire. J Clin Invest 2008 May;118(5):1712-26.http://www.ncbi.nlm.nih.gov/pubmed/18414681?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(7)
Chen Z, Herman AE, Matos M, Mathis D, Benoist
C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med 2005
Nov 21;202(10):1387-97.http://www.ncbi.nlm.nih.gov/pubmed/16301745?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(8)
Mordes JP, Bortell R, Blankenhorn EP,
Rossini AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment,
and autoimmunity. ILAR J 2004;45(3):278-91.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15229375
(9)
Leiter EH. Mice with targeted gene
disruptions or gene insertions for diabetes research: problems, pitfalls, and
potential solutions. diabetol 2002 Mar;45(3):296-308.http://www.ncbi.nlm.nih.gov/pubmed/11914735?ordinalpos=7&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(10)
Roep BO, Atkinson M, von Herrath M.
Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1
diabetes. Nat Rev Immunol 2004 Dec;4(12):989-97.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15573133
(11)
Anderson MS, Bluestone JA. The NOD
Mouse: A Model of Immune Dysregulation. Annu Rev Immunol 2004 Nov 29.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15568997
(12)
Devendra D, Liu E, Eisenbarth GS. Type 1
diabetes: recent developments. Brit Med J 2004;328(7442):750-4.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=15044291&query_hl=1&itool=pubmed_docsum
(13)
Eisenbarth GS, Gottlieb PA. Autoimmune
polyendocrine syndromes. N Engl J Med 2004 May 13;350(20):2068-79.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=15141045&query_hl=3&itool=pubmed_docsum
(14)
Homann D, von Herrath M. Regulatory T
cells and type 1 diabetes. Clin Immunol 2004 Sep;112(3):202-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15308110
(15)
Mathis D, Benoist C. Back to central
tolerance. Immunity 2004 May;20(5):509-16.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15142520
(16)
Panagiotopoulos C, Trudeau JD, Tan R.
T-cell epitopes in type 1 diabetes. Curr Diab Rep 2004 Apr;4(2):87-94.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15035967
(17)
Polychronakos C. Animal models of
spontaneous autoimmune diabetes: notes on their relevance to the human disease.
Curr Diab Rep 2004 Apr;4(2):151-4.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15035976
(18)
Ramsdell F. Foxp3 and natural regulatory
T cells: key to a cell lineage? Immunity 2003 Aug;19(2):165-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12932350
(19)
King M, Pearson T, Shultz LD, Leif J,
Bottino R, Trucco M, et al. Development of new-generation HU-PBMC-NOD/SCID mice
to study human islet alloreactivity. Ann N Y Acad Sci 2007 Apr;1103:90-3.
Epub@2007 Mar 21.:90-3.http://www.ncbi.nlm.nih.gov/pubmed/17376822?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(20)
Pearson T, Greiner DL, Shultz LD.
Creation of "humanized" mice to study human immunity. Curr Protoc
Immunol 2008 May;Chapter 15:Unit 15.21.:Unit.http://www.ncbi.nlm.nih.gov/pubmed/18491294?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(21)
Enee E, Martinuzzi E, Blancou P, Bach
JM, Mallone R, van Endert P. Equivalent specificity of peripheral blood and
islet-infiltrating CD8+ T lymphocytes in spontaneously diabetic HLA-A2
transgenic NOD mice. J Immunol 2008 Apr 15;180(8):5430-8.http://www.ncbi.nlm.nih.gov/pubmed/18390725?ordinalpos=19&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(22)
Kasprowicz DJ, Smallwood PS, Tyznik AJ,
Ziegler SF. Scurfin (FoxP3) controls T-dependent immune responses in vivo
through regulation of CD4+ T cell effector function. J Immunol 2003 Aug
1;171(3):1216-23.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12874208
(23)
Anderson MS, Venanzi ES, Klein L, Chen
Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow
within the thymus by the aire protein. Science 2002 Nov 15;298(5597):1395-401.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12376594
(24)
Liston A, Lesage S, Wilson J, Peltonen
L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat
Immunol 2003 Apr;4(4):350-4.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12612579
(25)
Yang Y, Santamaria P. Lessons on
autoimmune diabetes from animal models. Clin Sci (Lond) 2006 Jun;110(6):627-39.http://www.ncbi.nlm.nih.gov/pubmed/16689681?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(26)
Roep BO. Are insights gained from NOD
mice sufficient to guide clinical translation? Another inconvenient truth. Ann
N Y Acad Sci 2007 Apr;1103:1-10. Epub@2007 Mar 21.:1-10.http://www.ncbi.nlm.nih.gov/pubmed/17376838?ordinalpos=31&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(27)
Shoda LK, Young DL, Ramanujan S, Whiting
CC, Atkinson MA, Bluestone JA, et al. A comprehensive review of interventions
in the NOD mouse and implications for translation. Immunity 2005
Aug;23(2):115-26.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=16111631
(28)
Herold KC, Hagopian W, Auger JA,
Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in
new-onset type 1 diabetes mellitus. N Engl J Med 2002 May
30;346(22):1692-8.PM:12037148
(29)
Eisenbarth SC, Flavell RA. Innate
instruction of adaptive immunity revisited: the inflammasome. EMBO Mol Med 2009
May;1(2):92-8.PM:20049709
(30)
Moriyama H, Abiru N, Paronen J, Sikora
K, Liu E, Miao D, et al. Evidence for a primary islet autoantigen
(preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse.
Proc Natl Acad Sci U S A 2003 Sep 2;100(18):10376-81.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=Abstract&list_uids=12925730
(31)
Perone MJ, Bertera S, Shufesky WJ,
Divito SJ, Montecalvo A, Mathers AR, et al. Suppression of autoimmune diabetes
by soluble galectin-1. J Immunol 2009 Mar 1;182(5):2641-53.PM:19234158
(32)
Homann D, Eisenbarth GS. An immunologic
homunculus for type 1 diabetes. J Clin Invest 2006 May;116(5):1212-5.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=16670763
(33)
Santamaria P. The long and winding road
to understanding and conquering type 1 diabetes. Immunity 2010 Apr
23;32(4):437-45.PM:20412754
(34)
Eisenbarth GS. Banting Lecture 2009: An
Unfinished Journey: Molecular Pathogenesis to Prevention of Type 1A Diabetes.
diab 2010 Apr;59(4):759-74.PM:20350969
(35)
Singh B, Delovitch TL. Immune mechanisms
that regulate susceptibility to autoimmune type I diabetes. Clin Rev Allergy
Immunol 2000 Dec;19(3):247-64.http://www.ncbi.nlm.nih.gov/pubmed/11138408?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(36)
Zhang L, Nakayama M, Eisenbarth GS.
Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol 2008 Jan 4;.http://www.ncbi.nlm.nih.gov/pubmed/18178393?ordinalpos=28&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(37)
Driver JP, Serreze DV, Chen YG. Mouse
models for the study of autoimmune type 1 diabetes: a NOD to similarities and
differences to human disease. Semin Immunopathol 2011
Jan;33(1):67-87.PM:20424843
(38)
Chaparro RJ, DiLorenzo TP. An update on
the use of NOD mice to study autoimmune (Type 1) diabetes. Expert Rev Clin
Immunol 2010 Nov;6(6):939-55.PM:20979558
(39)
Giarratana N, Penna G, Adorini L. Animal
models of spontaneous autoimmune disease: type 1 diabetes in the nonobese
diabetic mouse. Methods Mol Biol 2007;380:285-311.:285-311.http://www.ncbi.nlm.nih.gov/pubmed/17876100?ordinalpos=17&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(40)
Ablamunits V, Sherry NA, Kushner JA,
Herold KC. Autoimmunity and beta cell regeneration in mouse and human type 1
diabetes: the peace is not enough. Ann N Y Acad Sci 2007 Apr;1103:19-32.
Epub@2007 Mar 21.:19-32.http://www.ncbi.nlm.nih.gov/pubmed/17376841?ordinalpos=30&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(41)
Di Lorenzo TP, Peakman M, Roep BO.
Translational mini-review series on type 1 diabetes: Systematic analysis of T
cell epitopes in autoimmune diabetes. Clin Exp Immunol 2007 Apr;148(1):1-16.http://www.ncbi.nlm.nih.gov/pubmed/17349009?ordinalpos=34&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(42)
Stadinski B, Kappler J, Eisenbarth GS.
Molecular targeting of islet autoantigens. Immunity 2010 Apr
23;32(4):446-56.PM:20412755
(43)
Fierabracci A. Peptide immunotherapies
in Type 1 diabetes: lessons from animal models. Curr Med Chem
2011;18(4):577-86.PM:21143110
(44)
Wicker LS, Clark J, Fraser HI, Garner
VE, Gonzalez-Munoz A, Healy B, et al. Type 1 diabetes genes and pathways shared
by humans and NOD mice. J Autoimmun 2005;25 Suppl:29-33. Epub@2005 Oct
28.:29-33.http://www.ncbi.nlm.nih.gov/pubmed/16257508?ordinalpos=74&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(45)
Suri A, Unanue ER. The Murine
Diabetogenic Class II Histocompatibility Molecule I-A(g7): Structural and
Functional Properties and Specificity of Peptide Selection. Adv Immunol
2005;88:235-65.:235-65.http://www.ncbi.nlm.nih.gov/pubmed/16227092?ordinalpos=76&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(46)
Haskins K. Pathogenic T-cell clones in
autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol
2005;87:123-62.:123-62.http://www.ncbi.nlm.nih.gov/pubmed/16102573?ordinalpos=88&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(47)
Chatenoud L, Bach JF. Regulatory T cells
in the control of autoimmune diabetes: the case of the NOD mouse. Int Rev
Immunol 2005 May;24(3-4):247-67.http://www.ncbi.nlm.nih.gov/pubmed/16036377?ordinalpos=93&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(48)
Hussain S, Wagner M, Ly D, Delovitch TL.
Role of regulatory invariant CD1d-restricted natural killer T-cells in
protection against type 1 diabetes. Immunol Res 2005;31(3):177-88.http://www.ncbi.nlm.nih.gov/pubmed/15888910?ordinalpos=99&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(49)
Leiter EH. Nonobese diabetic mice and
the genetics of diabetes susceptibility. Curr Diab Rep 2005 Apr;5(2):141-8.http://www.ncbi.nlm.nih.gov/pubmed/15794919?ordinalpos=101&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(50)
Anderson MS, Bluestone JA. The NOD
mouse: a model of immune dysregulation. Annu Rev Immunol
2005;23:447-85.:447-85.http://www.ncbi.nlm.nih.gov/pubmed/15771578?ordinalpos=103&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(51)
Babad J, Geliebter A, DiLorenzo TP.
T-cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes.
Immunology 2010 Dec;131(4):459-65.PM:21039471
(52)
Makino S, Kunimoto K, Muraoka Y,
Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of
mice. Exp Anim 1980;29:1-13.http://www.ncbi.nlm.nih.gov/pubmed/6995140?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(53)
Kanazawa Y, Komeda K, Sato S, Mori S,
Akanuma K, Takaku F. Non-obese-diabetic mice: immune mechanisms of pancreatic
B-cell destruction. diabetol 1984;27:113-5.http://www.ncbi.nlm.nih.gov/pubmed/6383913?ordinalpos=15&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(54)
Wicker LS, Miller BJ, Coker LZ, McNally
SE, Scott S, Mullen Y, et al. Genetic control of diabetes and insulitis in the
nonobese diabetic (NOD) mouse. J Exp Med 1987;165:1639-54.http://www.ncbi.nlm.nih.gov/pubmed/3585250?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(55)
Maruyama T, Takei I, Yanagawa T,
Takahashi T, Asaba Y, Kataoka K, et al. Insulin autoantibodies in non-obese
diabetic (NOD) mice and streptozotocin induced diabetic mice. Diabetes Res
1988;7(2):93-6.http://www.ncbi.nlm.nih.gov/pubmed/3293880?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(56)
Ziegler AG, Vardi P, Ricker AT, Hattori
M, Soeldner JS, Eisenbarth GS. Radioassay determination of insulin
autoantibodies in NOD mice. Correlation with increased risk of progression to
overt diabetes. diab 1989;38(3):358-63.http://www.ncbi.nlm.nih.gov/pubmed/2917700?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(57)
Gottlieb PA, Eisenbarth GS. Mouse and
man: multiple genes and multiple autoantigens in the aetiology of type I DM and
related autoimmune disorders. J Autoimmun 1996;9:277-81.http://www.ncbi.nlm.nih.gov/pubmed/8738974?ordinalpos=5&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(58)
Ott PA, Anderson MR, Tary-Lehmann M,
Lehmann PV. CD4+CD25+ regulatory T cells control the progression from
periinsulitis to destructive insulitis in murine autoimmune diabetes. Cell
Immunol 2005 May;235(1):1-11.http://www.ncbi.nlm.nih.gov/pubmed/16122720?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(59)
Todd JA, Aitman TJ, Cornall RJ, Ghosh S,
Hall JR, Hearne CM, et al. Genetic analysis of autoimmune type I diabetes
mellitus in mice. Nature 1991;351(6327):542-7.http://www.ncbi.nlm.nih.gov/pubmed/1675432?ordinalpos=17&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(60)
Oldstone MBA. Prevention of type 1
diabetes in nonobese diabetic mice by virus infection. Science 1988;239:500-2.http://www.ncbi.nlm.nih.gov/pubmed/3277269?ordinalpos=14&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(61)
Wilberz S, Partke HJ, Dagnaes-Hansen F,
Herberg L. Persistent MHV (mouse hepatitis virus) infection reduces the
incidence of diabetes mellitus in non-obese diabetic mice. diabetol
1991;34(1):2-5.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=1647335
(62)
Atkinson MA, Leiter EH. The NOD mouse
model of type 1 diabetes: as good as it gets? Nat Med 1999 Jun;5(6):601-4.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=10371488
(63)
Rainbow DB, Esposito L, Howlett SK,
Hunter KM, Todd JA, Peterson LB, et al. Commonality in the genetic control of
Type 1 diabetes in humans and NOD mice: variants of genes in the IL-2 pathway
are associated with autoimmune diabetes in both species. Biochem Soc Trans 2008
Jun 1;36(3):312-5.http://www.ncbi.nlm.nih.gov/pubmed/18481948?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(64)
Hattori M, Buse JB, Jackson RA, Glimcher
L, Dorf ME, Minami M, et al. The NOD mouse: recessive diabetogenic gene within
the major histocompatibility complex. Science 1986;231(4739):733-5.http://www.ncbi.nlm.nih.gov/pubmed/3003909?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(65)
Todd JA, Bell JI, McDevitt HO. HLA-DQB
gene contributes to susceptibility and resistance to insulin-dependent diabetes
mellitus. Nature 1987;329(6140):599-604.http://www.ncbi.nlm.nih.gov/pubmed/3309680?ordinalpos=9&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(66)
Wicker LS, Todd JA, Peterson LB. Genetic
control of autoimmune diabetes in the NOD mouse. Ann Rev Immunol
1995;13:179-200.http://www.ncbi.nlm.nih.gov/pubmed/7612220?ordinalpos=18&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(67)
Todd JA, Acha-Orbea H, Bell JI, Chao N,
Fronek Z, Jacob CO, et al. A molecular basis for MHC class II associated
autoimmunity. Science 1988;240(4855):1003-9.http://www.ncbi.nlm.nih.gov/pubmed/3368786?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(68)
Lund T, O'Reilly L, Hutchings P,
Kanagawa O, Simpson E, Gravely R, et al. Prevention of insulin-dependent
diabetes mellitus in non-obese diabetic mice by transgenes encoding modified
I-A beta-chain or normal I-E alpha-chain. Nature 1990;345(6277):727-9.http://www.ncbi.nlm.nih.gov/pubmed/2163026?ordinalpos=12&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(69)
Inoue K, Ikegami H, Fujisawa T, Noso S,
Nojima K, Babaya N, et al. Allelic variation in class I K gene as candidate for
a second component of MHC-linked susceptibility to type 1 diabetes in non-obese
diabetic mice. diabetol 2004 Apr;47(4):739-47.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15298352
(70)
Fujisawa T, Ikegami H, Noso S, Yamaji K,
Nojima K, Babaya N, et al. MHC-linked susceptibility to type 1 diabetes in the
NOD mouse: further localization of Idd16 by subcongenic analysis. Ann N Y Acad
Sci 2006 Oct;1079:118-21.:118-21.http://www.ncbi.nlm.nih.gov/pubmed/17130541?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(71)
Yamaji K, Ikegami H, Fujisawa T, Noso S,
Nojima K, Babaya N, et al. Contribution of class III MHC to susceptibility to
type 1 diabetes in the NOD mouse. Ann N Y Acad Sci 2006 Oct;1079:114-7.:114-7.http://www.ncbi.nlm.nih.gov/pubmed/17130540?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(72)
Mohan JF, Levisetti MG, Calderon B,
Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin
peptides generated within the islets of Langerhans in autoimmune diabetes. Nat
Immunol 2010 Apr;11(4):350-4.PM:20190756
(73)
Ueda H, Howson JM, Esposito L, Heward J,
Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4
with susceptibility to autoimmune disease. Nature 2003 Apr 30;423(6939):506-11.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12724780
(74)
Yamanouchi J, Rainbow D, Serra P,
Howlett S, Hunter K, Garner VE, et al. Interleukin-2 gene variation impairs
regulatory T cell function and causes autoimmunity. Nat Genet 2007 Feb 4;.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=17277778&query_hl=15&itool=pubmed_docsum
(75)
Hamilton-Williams EE, Serreze DV,
Charlton B, Johnson EA, Marron MP, Mullbacher A, et al. Transgenic rescue
implicates beta2-microglobulin as a diabetes susceptibility gene in nonobese
diabetic (NOD) mice. Proc Natl Acad Sci U S A 2001 Sep 25;98(20):11533-8.http://www.ncbi.nlm.nih.gov/pubmed/11572996?ordinalpos=12&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(76)
Todd JA, Wicker LS. Genetic protection
from the inflammatory disease type 1 diabetes in humans and animal models.
Immunity 2001 Sep;15(3):387-95.http://www.ncbi.nlm.nih.gov/pubmed/11567629?ordinalpos=10&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(77)
Robles DT, Eisenbarth GS, Dailey NJM,
Peterson LB, Wicker LS. Insulin autoantibodies are associated with islet
inflammation but not always related to diabetes progression in NOD congenic
mice. diab 2002;52(3):882-6.http://www.ncbi.nlm.nih.gov/pubmed/12606534?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(78)
Encinas JA, Kuchroo VK. Mapping and
identification of autoimmunity genes. Curr Opin Immunol 2000 Dec;12(6):691-7.http://www.ncbi.nlm.nih.gov/pubmed/11102774?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(79)
McDuffie M. Genetics of autoimmune
diabetes in animal models. Curr Opin Immunol 1998 Dec;10(6):704-9.http://www.ncbi.nlm.nih.gov/pubmed/9914220?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(80)
Wakeland E, Morel L, Achey K, Yui M,
Longmate J. Speed congenics: a classic technique in the fast lane (relatively
speaking). Immunol Today 1997;18:472-7.http://www.ncbi.nlm.nih.gov/pubmed/9357138?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(81)
Brook FA, Evans EP, Lord CJ, Lyons PA,
Rainbow DB, Howlett SK, et al. The derivation of highly germline-competent
embryonic stem cells containing NOD-derived genome. diab 2003 Jan;52(1):205-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12502514
(82)
Kawabata Y, Ikegami H, Fujisawa T, Noso
S, Asano K, Hiromine Y, et al. A second component of HLA-linked susceptibility
to type 1 diabetes maps to class I region. Ann N Y Acad Sci 2006
Oct;1079:278-84.:278-84.http://www.ncbi.nlm.nih.gov/pubmed/17130566?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(83)
Ikegami H, Fujisawa T, Makino S, Ogihara
T. Congenic mapping and candidate sequencing of susceptibility genes for Type 1
diabetes in the NOD mouse. Ann N Y Acad Sci 2003 Nov;1005:196-204.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14679059
(84)
Takaki T, Lieberman SM, Holl TM, Han B,
Santamaria P, Serreze DV, et al. Requirement for both H-2Db and H-2Kd for the
induction of diabetes by the promiscuous CD8+ T cell clonotype AI4. J Immunol
2004 Aug 15;173(4):2530-41.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15294969
(85)
Acha-Orbea H, McDevitt HO. The first
external domain of the nonobese diabetic mouse class II I-A chain is unique.
Proc Natl Acad Sci USA 1987;84(8):2435-9.http://www.ncbi.nlm.nih.gov/pubmed/2882518?ordinalpos=15&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(86)
Nepom GT, Erlich H. MHC class-II
molecules and autoimmunity. Ann Rev Immunol 1991;9:493-525.http://www.ncbi.nlm.nih.gov/pubmed/1910687?ordinalpos=53&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(87)
Miyazaki T, Uno M, Uehira M, Kikutani H,
Kishimoto T, Kimoto M, et al. Direct evidence for the contribution of the
unique I-ANOD to the development of insulitis in non-obese diabetic mice.
Nature 1990 Jun 21;345(6277):722-4
(88)
Nishimoto H, Kikutani H, Yamamura K,
Kishimoto T. Prevention of autoimmune insulitis by expression of I-E molecules
in NOD mice. Nature 1987;328(6129):432-4
(89)
Wherrett DK, Singer SM, McDevitt HO.
Reduction in diabetes incidence in an I-Ag7 transgenic nonobese
diabetic mouse line. diab 1997;46(12):1970-4.http://www.ncbi.nlm.nih.gov/pubmed/9392482?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(90)
Ikegami H, Makino S, Harada M,
Eisenbarth GS, Hattori M. The cataract Shionogi mouse,a sister non-obese
diabetic mouse similar class II but different class I gene products. diabetol
1988;31(4):254-8.http://www.ncbi.nlm.nih.gov/pubmed/3133269?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(91)
Ikegami H, Eisenbarth GS, Hattori M.
Major histocompatibility complex-linked diabetogenic gene of the nonobese
diabetic mouse. Analysis of genomic DNA amplified by polymerase chain reaction.
J Clin Invest 1990;85(1):18-24.http://www.ncbi.nlm.nih.gov/pubmed/2295694?ordinalpos=17&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(92)
Wicker LS, Miller BJ, Fischer PA,
Pressey A, Peterson LB. Genetic control of diabetes and insulitis in the
nonobese diabetic mouse. Pedigree analysis of a diabetic H-2nod/b heterozygote.
J Immunol 1989 Feb 1;142(3):781-4.http://www.ncbi.nlm.nih.gov/pubmed/2643662?ordinalpos=43&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(93)
Corper AL, Stratmann T, Apostolopoulos
V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering
the link between I-Ag7 and autoimmune diabetes. Science 2000 Apr
21;288(5465):505-11.http://www.ncbi.nlm.nih.gov/pubmed/10775108?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(94)
Latek RR, Suri A, Petzold SJ, Nelson CA,
Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and
presentation by the type I diabetes-associated MHC class II molecule of NOD
mice. Immunity 2000 Jun;12(6):699-710.http://www.ncbi.nlm.nih.gov/pubmed/10894169?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(95)
Carrasco-Marin E, Kanagawa O, Unanue ER.
Insights into the chemistry and biology of the I-Ag7 class II
molecule. Res Immunol 1997;148:291-301.http://www.ncbi.nlm.nih.gov/pubmed/9352592?ordinalpos=31&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(96)
Kanagawa O, Martin SM, Vaupel BA,
Carrasco-Marin E, Unanue ER. Autoreactivity of T cells from nonobese diabetic
mice: an I-Ag7-dependent reaction. Proc Natl Acad Sci U S A 1998 Feb
17;95(4):1721-4.http://www.ncbi.nlm.nih.gov/pubmed/9465083?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(97)
Duarte N, Lundholm M, Holmberg D. The
Idd6.2 diabetes susceptibility region controls defective expression of the Lrmp
gene in nonobese diabetic (NOD) mice. Immunogenetics 2007 May;59(5):407-16.http://www.ncbi.nlm.nih.gov/pubmed/17353998?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(98)
Schmidt D, Verdaguer J, Averill N,
Santamaria P. A mechanism for the major histocompatibility complex-linked
resistance to autoimmunity. J Exp Med 1997;186:1059-75.http://www.ncbi.nlm.nih.gov/pubmed/9314555?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(99)
Ridgway WM, Ito H, Fasso M, Yu C,
Fathman CG. Analysis of the role of variation of major histocompatibility
complex class II expression on nonobese diabetic (NOD) peripheral T cell
response. J Exp Med 1998 Dec 21;188(12):2267-75
(100)
van Rijn JC, Grote FK, Oostdijk W, Wit
JM. Short stature and the probability of coeliac disease, in the absence of
gastrointestinal symptoms. Arch Dis Child 2004 Sep;89(9):882-3.http://www.ncbi.nlm.nih.gov/pubmed/15321874?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(101)
Kanagawa O, Vaupel BA, Xu G, Unanue ER,
Katz JD. Thymic positive selection and peripheral activation of islet
antigen-specific T cells: separation of two diabetogenic steps by an I-A(g7)
class II MHC beta-chain mutant. J Immunol 1998 Nov 1;161(9):4489-92.http://www.ncbi.nlm.nih.gov/pubmed/9794372?ordinalpos=18&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(102)
Suri A, Katz JD. Dissecting the role of
CD4+ T cells in autoimmune diabetes through the use of TCR transgenic mice.
Immunol Rev 1999 Jun;169:55-65.http://www.ncbi.nlm.nih.gov/pubmed/10450508?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(103)
Nakayama M, Beilke JN, Jasinski JM,
Kobayashi M, Miao D, Li M, et al. Priming and effector dependence on insulin
B:9-23 peptide in NOD islet autoimmunity. J Clin Invest 2007 Jul
2;117(7):1835-43.http://www.ncbi.nlm.nih.gov/sites/entrez?Db=pubmed&Cmd=ShowDetailView&TermToSearch=17607359&ordinalpos=20&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(104)
Thebault-Baumont K, Dubois-LaForgue D,
Krief P, Briand JP, Halbout P, Vallon-Geoffroy K, et al. Acceleration of type 1
diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest 2003 Mar
15;111(6):851-7.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12639991
(105)
Levisetti MG, Suri A, Petzold SJ, Unanue
ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak
MHC-binding segment in more than one form. J Immunol 2007 May
15;178(10):6051-7.http://www.ncbi.nlm.nih.gov/pubmed/17475829?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(106)
Michels AW, Nakayama M. 2The
anti-insulin trimolecular complex in type 1 diabetes. Curr Opin Endocrinol
Diabetes Obes 2010 May 19.PM:20489610
(107)
Kobayashi M, Jasinski J, Liu E, Li M,
Miao D, Zhang L, et al. Conserved T cell receptor alpha-chain induces insulin
autoantibodies. Proc Natl Acad Sci U S A 2008 Jul 22;105(29):10090-4
(108)
Nikoopour E, Sandrock C, Huszarik K,
Krougly O, Lee-Chan E, Masteller EL, et al. Cutting Edge: Vasostatin-1-Derived
Peptide ChgA29-42 Is an Antigenic Epitope of Diabetogenic BDC2.5 T Cells in
Nonobese Diabetic Mice. J Immunol 2011 Apr 1;186(7):3831-5.PM:21357258
(109) Ikegami H, Jackson RA, Makino S, Watt DE,
Eisenbarth GS, Hattori M. Homozygosity for two genes (H-2: chromosome 17 and
Thy-1: chromosome 9) linked to development of type I diabetes of the NOD mouse.
Clin Res 34, 683A. 1986.
Ref Type: Abstract
(110)
Lyons PA, Hancock WW, Denny P, Lord CJ,
Hill NJ, Armitage N, et al. The NOD Idd9 genetic interval influences the
pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and
Cd137. Immunity 2000 Jul;13(1):107-15.http://www.ncbi.nlm.nih.gov/pubmed/10933399?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(111)
Podolin PL, Wilusz MB, Cubbon RM,
Pajvani U, Lord CJ, Todd JA, et al. Differential glycosylation of interleukin
2, the molecular basis for the NOD Idd3 type 1 diabetes gene? Cytokine 2000
May;12(5):477-82.http://www.ncbi.nlm.nih.gov/pubmed/10857762?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(112)
Eisenbarth SC, Homann D. "Primer
Immunology and Autoimmunity". Type I Diabetes: Molecular, Cellular and
Clinical Immunology 2006 Nov.http://barbaradaviscenter.org/
(113)
Ikegami H, Fujisawa T, Sakamoto T,
Makino S, Ogihara T. Idd1 and Idd3 are necessary but not sufficient for
development of type 1 diabetes in NOD mouse. Diabetes Res Clin Pract 2004
Dec;66 Suppl 1:S85-S90.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15563987
(114)
del Rio R, Noubade R, Subramanian M,
Saligrama N, Diehl S, Rincon M, et al. SNPs upstream of the minimal promoter
control IL-2 expression and are candidates for the autoimmune
disease-susceptibility locus Aod2/Idd3/Eae3. Genes Immun 2008 Mar;9(2):115-21.http://www.ncbi.nlm.nih.gov/pubmed/18200031?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(115)
Ghosh S, Palmer SM, Rodrigues NR,
Cordell HJ, Hearne CM, Cornall RJ, et al. Polygenic control of autoimmune
diabetes in nonobese diabetic mice. Nat Genet 1993;4:404-9.http://www.ncbi.nlm.nih.gov/sites/entrez?orig_db=PubMed&db=pubmed&cmd=Search&term=4%5Bvolume%5D%20AND%20404%5Bpage%5D%20AND%201993%5Bpdat%5D
(116)
McDuffie M, Maybee NA, Keller SR,
Stevens BK, Garmey JC, Morris MA, et al. Nonobese diabetic (NOD) mice congenic
for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune
diabetes. Diabetes 2008 Jan;57(1):199-208.http://www.ncbi.nlm.nih.gov/pubmed/17940120?ordinalpos=121&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(117)
Davoodi-Semiromi A, McDuffie M,
Litherland S, Clare-Salzler M. Truncated pStat5B is associated with the Idd4
locus in NOD mice. Biochem Biophys Res Commun 2007 May 11;356(3):655-61.http://www.ncbi.nlm.nih.gov/pubmed/17382905?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(118)
Hill NJ, Lyons PA, Armitage N, Todd JA,
Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and
overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans [In Process
Citation]. diab 2000 Oct;49(10):1744-7.http://www.ncbi.nlm.nih.gov/pubmed/11016460?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(119)
Fox CJ, Paterson AD, Mortin-Toth SM,
Danska JS. Two genetic loci regulate T cell-dependent islet inflammation and
drive autoimmune diabetes pathogenesis. Am J Hum Genet 2000 Jul;67(1):67-81.http://www.ncbi.nlm.nih.gov/pubmed/10848492?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(120)
Cordell HJ, Todd JA, Hill NJ, Lord CJ,
Lyons PA, Peterson LB, et al. Statistical modeling of interlocus interactions
in a complex disease: rejection of the multiplicative model of epistasis in
type 1 diabetes. Genetics 2001 May;158(1):357-67.http://www.ncbi.nlm.nih.gov/pubmed/11333244?ordinalpos=19&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(121)
Wicker LS, Chamberlain G, Hunter K,
Rainbow D, Howlett S, Tiffen P, et al. Fine mapping, gene content, comparative
sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for
Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol 2004 Jul
1;173(1):164-73.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15210771
(122)
Greve B, Vijayakrishnan L, Kubal A,
Sobel RA, Peterson LB, Wicker LS, et al. The diabetes susceptibility locus
Idd5.1 on mouse chromosome 1 regulates ICOS expression and modulates murine
experimental autoimmune encephalomyelitis. J Immunol 2004 Jul 1;173(1):157-63.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15210770
(123)
Araki M, Chung D, Liu S, Rainbow DB,
Chamberlain G, Garner V, et al. Genetic evidence that the differential
expression of the ligand-independent isoform of CTLA-4 is the molecular basis
of the Idd5.1 type 1 diabetes region in nonobese diabetic mice. J Immunol 2009
Oct 15;183(8):5146-57.PM:19783679
(124)
Colucci F, Bergman ML, Penha-Goncalves
C, Cilio CM, Holmberg D. Apoptosis resistance of nonobese diabetic peripheral
lymphocytes linked to the Idd5 diabetes susceptibility region. Proc Natl Acad
Sci U S A 1997 Aug 5;94(16):8670-4.http://www.ncbi.nlm.nih.gov/pubmed/9238035?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(125)
Bergman ML, Cilio CM, Penha-Goncalves C,
Lamhamedi-Cherradi SE, Lofgren A, Colucci F, et al. CTLA-4-/- mice display T
cell-apoptosis resistance resembling that ascribed to autoimmune-prone
non-obese diabetic (NOD) mice. J Autoimmun 2001 Mar;16(2):105-13.http://www.ncbi.nlm.nih.gov/pubmed/11247636?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(126)
Motta V, Lejon K, Holmberg D. The NOD
allele of the Idd5 locus on chromosome 1 mediates a non-cell-autonomous defect
in negative selection of T cells. J Autoimmun 2007 Jun;28(4):216-23.http://www.ncbi.nlm.nih.gov/pubmed/17449224?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(127)
Irie J, Reck B, Wu Y, Wicker LS, Howlett
S, Rainbow D, et al. Genome-wide microarray expression analysis of CD4+ T Cells
from nonobese diabetic congenic mice identifies Cd55 (Daf1) and Acadl as
candidate genes for type 1 diabetes. J Immunol 2008 Jan 15;180(2):1071-9.http://www.ncbi.nlm.nih.gov/pubmed/18178847?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(128)
Carnaud C, Gombert J, Donnars O, Garchon
H, Herbelin A. Protection against diabetes and improved NK/NKT cell performance
in NOD.NK1.1 mice congenic at the NK complex. J Immunol 2001 Feb
15;166(4):2404-11.http://www.ncbi.nlm.nih.gov/pubmed/11160299?ordinalpos=5&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(129)
Laloux V, Beaudoin L, Jeske D, Carnaud
C, Lehuen A. NK T cell-induced protection against diabetes in V alpha 14-J
alpha 281 transgenic nonobese diabetic mice is associated with a Th2 shift
circumscribed regionally to the islets and functionally to islet autoantigen. J
Immunol 2001 Mar 15;166(6):3749-56.http://www.ncbi.nlm.nih.gov/pubmed/11238616?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(130)
Sharif S, Arreaza GA, Zucker P, Mi QS,
Sondhi J, Naidenko OV, et al. Activation of natural killer T cells by
alpha-galactosylceramide treatment prevents the onset and recurrence of
autoimmune Type 1 diabetes. Nat Med 2001 Sep;7(9):1057-62.http://www.ncbi.nlm.nih.gov/pubmed/11533711?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(131)
Bergman ML, Penha-Goncalves C, Lejon K,
Holmberg D. Low rate of proliferation in immature thymocytes of the non-obese
diabetic mouse maps to the Idd6 diabetes susceptibility region. diabetol 2001
Aug;44(8):1054-61.http://www.ncbi.nlm.nih.gov/pubmed/11484085?ordinalpos=24&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(132)
Vallois D, Grimm CH, Avner P, Boitard C,
Rogner UC. The type 1 diabetes locus Idd6 controls TLR1 expression. J Immunol
2007 Sep 15;179(6):3896-903.http://www.ncbi.nlm.nih.gov/pubmed/17785827?ordinalpos=156&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(133)
Serreze DV, Choisy-Rossi CM, Grier AE,
Holl TM, Chapman HD, Gahagan JR, et al. Through regulation of TCR expression
levels, an Idd7 region gene(s) interactively contributes to the impaired thymic
deletion of autoreactive diabetogenic CD8+ T cells in nonobese diabetic mice. J
Immunol 2008 Mar 1;180(5):3250-9.http://www.ncbi.nlm.nih.gov/pubmed/18292549?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(134)
Siegmund T, Armitage N, Wicker LS,
Peterson LB, Todd JA, Lyons PA. Analysis of the mouse CD30 gene: a candidate
for the NOD mouse type 1 diabetes locus Idd9.2. diab 2000 Sep;49(9):1612-6.http://www.ncbi.nlm.nih.gov/pubmed/10969850?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(135)
Ueno A, Wang J, Cheng L, Im JS, Shi Y,
Porcelli SA, et al. Enhanced early expansion and maturation of semi-invariant
NK T cells inhibited autoimmune pathogenesis in congenic nonobese diabetic
mice. J Immunol 2008 Nov 15;181(10):6789-96.PM:18981096
(136)
Rolf J, Motta V, Duarte N, Lundholm M,
Berntman E, Bergman ML, et al. The enlarged population of marginal
zone/CD1d(high) B lymphocytes in nonobese diabetic mice maps to diabetes
susceptibility region Idd11. Journal of Immunology 2005;174(8):4821-7.<Go to
ISI>://000228234600048
(137)
Rolf J, Motta V, Duarte N, Lundholm M,
Berntman E, Bergman ML, et al. Congenic nonobese diabetic mouse strains fail to
confirm linkage of a marginal zone B lymphocyte phenotype to the Idd11 locus on
chromosome 4 - Response. Journal of Immunology 2006;176(2):702.<Go to
ISI>://000234553800002
(138)
Heath VL, Moore NC, Parnell SM, Mason
DW. Intrathymic expression of genes involved in organ specific autoimmune
disease. J Autoimmun 1998 Aug;11(4):309-18.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=9776708
(139)
Yamaji K, Ikegami H, Fujisawa T, Noso S,
Nojima K, Babaya N, et al. Evidence for Cd101 but not Fcgr1 as candidate for
type 1 diabetes locus, Idd10. Biochem Biophys Res Commun 2005 Jun
3;331(2):536-42.http://www.ncbi.nlm.nih.gov/pubmed/15850792?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(140)
Podolin PL, Denny P, Lord CJ, Hill NJ,
Todd JA, Peterson LB, et al. Congenic mapping of the insulin-dependent diabetes
(Idd) gene, Idd10, localizes two genes mediating the Idd10 effect and
eliminates the candidate Fcgr1. J Immunol 1997 Aug 15;159(4):1835-43.http://www.ncbi.nlm.nih.gov/pubmed/9257847?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(141)
Chen YG, Driver JP, Silveira PA, Serreze
DV. Subcongenic analysis of genetic basis for impaired development of invariant
NKT cells in NOD mice. Immunogenetics 2007;59(9):705-12.<Go to
ISI>://000249503200002
(142)
Schmidt MB. Eine biglandulare Erkrankung
(Nebennieren und Schilddruse) bei Morbus Addisonii. Verh Dtsch Ges Pathol
1926;21:212-21
(143)
Liston A, Gray DH, Lesage S, Fletcher
AL, Wilson J, Webster KE, et al. Gene dosage--limiting role of Aire in thymic
expression, clonal deletion, and organ-specific autoimmunity. J Exp Med 2004 Oct
18;200(8):1015-26.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15492124
(144)
Anderson MS. Autoimmune endocrine
disease. Curr Opin Immunol 2002 Dec;14(6):760-4.http://www.ncbi.nlm.nih.gov/pubmed/12413526?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(145)
Adams TE, Apert S, Hanahan D.
Non-tolerance and autoantibodies to a transgenic self-antigen expressed in
pancreatic beta cells. Nature 1987;325:223-8.http://www.ncbi.nlm.nih.gov/pubmed/3543686?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(146)
Hanahan D. Peripheral-antigen-expressing
cells in thymic medulla: factors in self- tolerance and autoimmunity. Curr Opin
Immunol 1998 Dec;10(6):656-62.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=9914224
(147)
Pugliese A. Genetics of type 1 diabetes.
Endocrinol Metab Clin North Am 2004 Mar;33(1):1-16, vii.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15053891
(148)
Cetani F, Barbesino G, Borsari S, Pardi
E, Cianferotti L, Pinchera A, et al. A novel mutation of the autoimmune
regulator gene in an Italian kindred with autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant
fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J
Clin Endocrinol Metab 2001 Oct;86(10):4747-52.http://www.ncbi.nlm.nih.gov/pubmed/11600535?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(149)
Gray DH, Gavanescu I, Benoist C, Mathis
D. Danger-free autoimmune disease in Aire-deficient mice. Proc Natl Acad Sci U
S A 2007 Nov 13;104(46):18193-8.http://www.ncbi.nlm.nih.gov/pubmed/17991771?ordinalpos=102&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(150)
Gambineri E, Torgerson TR, Ochs HD.
Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance
(IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a
critical regulator of T-cell homeostasis. Current Opinion in Rheumatology
2003;15(4):430-5.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12819471
(151)
Patel DD. Escape from tolerance in the
human X-linked autoimmunity-allergic disregulation syndrome and the Scurfy
mouse. J Clin Invest 2001;107(2):155-7.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11160129&query_hl=7
(152)
Chen Z, Benoist C, Mathis D. How defects
in central tolerance impinge on a deficiency in regulatory T cells. Proc Natl
Acad Sci U S A 2005 Oct 11;102(41):14735-40.http://www.ncbi.nlm.nih.gov/pubmed/16203996?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(153)
Griffiths MM, Encinas JA, Remmers EF,
Kuchroo VK, Wilder RL. Mapping autoimmunity genes. Curr Opin Immunol 1999
Dec;11(6):689-700.http://www.ncbi.nlm.nih.gov/pubmed/10631556?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(154)
Fairhurst AM, Wandstrat AE, Wakeland EK.
Systemic lupus erythematosus: multiple immunological phenotypes in a complex
genetic disease. Adv Immunol 2006;92:1-69.:1-69.http://www.ncbi.nlm.nih.gov/pubmed/17145301?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(155)
Bottini N, Musumeci L, Alonso A,
Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid
tyrosine phosphatase is associated with type I diabetes. Nat Genet 2004
Apr;36(4):337-8.http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15004560
(156)
Smyth D, Cooper JD, Collins JE, Heward
JM, Franklyn JA, Howson JM, et al. Replication of an Association Between the
Lymphoid Tyrosine Phosphatase Locus (LYP/PTPN22) With Type 1 Diabetes, and
Evidence for Its Role as a General Autoimmunity Locus. diab 2004
Nov;53(11):3020-3.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15504986
(157)
Velaga MR, Wilson V, Jennings CE, Owen
CJ, Herington S, Donaldson PT, et al. The Codon 620 Tryptophan Allele of the
Lymphoid Tyrosine Phosphatase (LYP) Gene Is a Major Determinant of Graves'
Disease. J Clin Endocrinol Metab 2004 Dec;89(11):5862-5.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15531553
(158)
Salomon B, Rhee L, Bour-Jordan H, Hsin
H, Montag A, Soliven B, et al. Development of spontaneous autoimmune peripheral
polyneuropathy in b7-2-deficient nod mice. J Exp Med 2001 Sep 3;194(5):677-84.http://www.ncbi.nlm.nih.gov/pubmed/11535635?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(159)
Winer S, Tsui H, Lau A, Song A, Li X,
Cheung RK, et al. Autoimmune islet destruction in spontaneous type 1 diabetes
is not beta-cell exclusive. Nat Med 2003 Feb;9(2):198-205.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12539039
(160)
Winer S, Astsaturov I, Cheung R,
Gunaratnam L, Kubiak V, Cortez MA, et al. Type I diabetes and multiple
sclerosis patients target islet plus central nervous system autoantigens;
nonimmunized nonobese diabetic mice can develop autoimmune encephalitis. J
Immunol 2001 Feb 15;166(4):2831-41.http://www.ncbi.nlm.nih.gov/pubmed/11160351?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(161)
Rivero VE, Cailleau C, Depiante-Depaoli
M, Riera CM, Carnaud C. Non-obese diabetic (NOD) mice are genetically
susceptible to experimental autoimmune prostatitis (EAP). J Autoimmun 1998
Dec;11(6):603-10.http://www.ncbi.nlm.nih.gov/pubmed/9878082?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(162)
Koarada S, Wu Y, Fertig N, Sass DA,
Nalesnik M, Todd JA, et al. Genetic control of autoimmunity: protection from
diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic
congenic strain. J Immunol 2004 Aug 15;173(4):2315-23.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15294944
(163)
Bendelac A, Carnaud C, Boitard C, Bach
JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy
neonates. Requirement for both L3T4+ and
Lyt-2+ T cells. J Exp Med 1987;166:823-32.http://www.ncbi.nlm.nih.gov/pubmed/3309126?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(164)
Wicker LS, Miller BJ, Mullen Y. Transfer
of autoimmune diabetes mellitus with splenocytes from non-obese diabetic (NOD)
mice. diab 1986;35(8):855-60.http://www.ncbi.nlm.nih.gov/pubmed/3525284?ordinalpos=7&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(165)
Charlton B, Mandel TE. Progression from
insulitis to Beta cell destruction in NOD mouse requires L3T4+T-lymphocytes.
diab 1988;37:1108-12.http://www.ncbi.nlm.nih.gov/pubmed/3292330?ordinalpos=7&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(166)
Serreze DV, Chapman HD, Varnum DS,
Hanson MS, Reifsnyder PC, Scott DR, et al. B lymphocytes are essential for the
initiation of T cell-mediated autoimmune diabetes: analysis of a new
"speed-congenic" stock of NOD.Igunull mice. J Exp Med
1996;184:2049-53.http://www.ncbi.nlm.nih.gov/pubmed/8920894?ordinalpos=11&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(167)
Greeley SA, Katsumata M, Yu L,
Eisenbarth GS, Moore DJ, Goodarzi H, et al. Elimination of maternally
transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med
2002 Apr;8(4):399-402.http://www.ncbi.nlm.nih.gov/pubmed/11927947?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(168)
Honkanen J, Nieminen JK, Gao R,
Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 immunity in human type 1
diabetes. J Immunol 2010 Aug 1;185(3):1959-67.PM:20592279
(169)
Sreenan S, Pick AJ, Levisetti M, Baldwin
AC, Pugh W, Polonsky KS. Increased b-cell proliferation and reduced mass before diabetes
onset in the nonobese diabetic mouse. diab 1999;48:989-96.http://www.ncbi.nlm.nih.gov/pubmed/10331402?ordinalpos=20&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(170)
Valorani MG, Hawa MI, Buckley LR,
Afeltra A, Cacciapaglia F, Pozzilli P. The natural history of insulin content
in the pancreas of female and male non-obese diabetic mouse: implications for
trials of diabetes prevention in humans. Diabetes Metab Res Rev 2004
Sep;20(5):394-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15343585
(171)
Alanentalo T, Hornblad A, Mayans S,
Karin NA, Sharpe J, Larefalk A, et al. Quantification and three-dimensional
imaging of the insulitis-induced destruction of beta-cells in murine type 1
diabetes. diab 2010 Jul;59(7):1756-64.PM:20393145
(172)
Nir T, Melton DA, Dor Y. Recovery from
diabetes in mice by beta cell regeneration. J Clin Invest 2007
Sep;117(9):2553-61.http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17786244
(173)
Meier JJ, Lin JC, Butler AE, Galasso R,
Martinez DS, Butler PC. Direct evidence of attempted beta cell regeneration in
an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia 2006
Aug;49(8):1838-44.http://www.ncbi.nlm.nih.gov/pubmed/16802132?ordinalpos=9&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(174)
Butler AE, Galasso R, Meier JJ, Basu R,
Rizza RA, Butler PC. Modestly increased beta cell apoptosis but no increased
beta cell replication in recent-onset type 1 diabetic patients who died of
diabetic ketoacidosis. diabetol 2007 Nov;50(11):2323-31.http://www.ncbi.nlm.nih.gov/pubmed/17805509?dopt=Citation
(175)
Perl S, Kushner JA, Buchholz BA, Meeker
AK, Stein GM, Hsieh M, et al. Significant human beta-cell turnover is limited
to the first three decades of life as determined by in vivo thymidine analog
incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010
Oct;95(10):E234-E239.PM:20660050
(176)
Keenan HA, Sun JK, Levine J, Doria A,
Aiello LP, Eisenbarth G, et al. Residual insulin production and pancreatic
ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. diab 2010
Nov;59(11):2846-53.PM:20699420
(177)
Sherry NA, Kushner JA, Glandt M,
Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune
therapy on beta-cell turnover in type 1 diabetes. Diabetes 2006
Dec;55(12):3238-45.http://www.ncbi.nlm.nih.gov/pubmed/17130466?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(178)
Haskins K, Atkinson M. Prediction of
Diabetes and Response to Therapy in Individual Animals: are there Lessons for
Man? Report on the JDF/Snow Mountain Meeting, 5-7 October 2000, Winter Park,
CO, USA. J Autoimmun 2001 Feb;16(1):15-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11221992
(179)
Bonifacio E, Atkinson M, Eisenbarth G,
Serreze D, Kay TW, Lee-Chan E, et al. International Workshop on Lessons from
Animal Models for Human Type 1 Diabetes: analyzing target autoantigens of
humoral immunity in nonobese diabetic mice. Ann N Y Acad Sci 2002 Apr;958:1-2.http://www.ncbi.nlm.nih.gov/pubmed/12021078?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(180)
Yu L, Eisenbarth GS, Bonifacio E, Thomas
J, Atkinson M, Wasserfall C. The Second Murine Autoantibody Workshop:
remarkable inter-laboratory concordance for radiobinding assays to identify
insulin autoantibodies in non-obese diabetic mice. NY Acad Sci 2003;100:1-12.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14679035
(181)
Moore DJ, Noorchashm H, Lin TH, Greeley
SA, Naji A. NOD B-cells are insufficient to incite T-cell-mediated anti-islet
autoimmunity. Diabetes 2005 Jul;54(7):2019-25.http://www.ncbi.nlm.nih.gov/pubmed/15983202?ordinalpos=12&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(182)
Noorchashm H, Noorchashm N, Kern J,
Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of
insulitis and sialitis in nonobese diabetic mice. diab 1997 Jun;46(6):941-6.http://www.ncbi.nlm.nih.gov/pubmed/9166663?ordinalpos=5&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(183)
Kendall PL, Yu G, Woodward EJ, Thomas
JW. Tertiary lymphoid structures in the pancreas promote selection of B
lymphocytes in autoimmune diabetes. J Immunol 2007 May 1;178(9):5643-51.http://www.ncbi.nlm.nih.gov/pubmed/17442947?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(184)
Hu CY, Rodriguez-Pinto D, Du W, Ahuja A,
Henegariu O, Wong FS, et al. Treatment with CD20-specific antibody prevents and
reverses autoimmune diabetes in mice. J Clin Invest 2007 Dec;117(12):3857-67.http://www.ncbi.nlm.nih.gov/pubmed/18060033?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(185)
Hoglund P, Mintern J, Waltzinger C,
Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by
developmentally regulated presentation of islet cell antigens in the pancreatic
lymph nodes. J Exp Med 1999 Jan 18;189(2):331-9.http://www.ncbi.nlm.nih.gov/pubmed/9892615?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(186)
Denis MC, Mahmood U, Benoist C, Mathis
D, Weissleder R. Imaging inflammation of the pancreatic islets in type 1
diabetes. Proc Natl Acad Sci U S A 2004 Aug 24;101(34):12634-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15304647
(187)
Lejon K, Fathman CG. Isolation of self
antigen-reactive cells from inflamed islets of nonobese diabetic mice using
CD4high expression as a marker. J Immunol 1999 Nov 15;163(10):5708-14.http://www.ncbi.nlm.nih.gov/pubmed/10553102?ordinalpos=17&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(188)
Gagnerault MC, Luan JJ, Lotton C,
Lepault F. Pancreatic lymph nodes are required for priming of beta cell
reactive T cells in NOD mice. J Exp Med 2002 Aug 5;196(3):369-77.http://www.ncbi.nlm.nih.gov/pubmed/12163565?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(189)
Poirot L, Benoist C, Mathis D. Natural
killer cells distinguish innocuous and destructive forms of pancreatic islet
autoimmunity. Proc Natl Acad Sci U S A 2004 May 25;101(21):8102-7.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15141080
(190)
Serreze DV, Gaskins HR, Leiter EH.
Defects in the differentiation and function of antigen presenting cells in
NOD/Lt mice. J Immunol 1993 Mar 15;150(6):2534-43.http://www.ncbi.nlm.nih.gov/pubmed/8450229?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(191)
Piganelli JD, Martin T, Haskins K.
Splenic macrophages from the NOD mouse are defective in the ability to present
antigen. diab 1998;47:1212-8.http://www.ncbi.nlm.nih.gov/pubmed/9703319?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(192)
Dahlen E, Hedlund G, Dawe K. Low CD86
expression in the nonobese diabetic mouse results in the impairment of both T
cell activation and CTLA-4 up-regulation. J Immunol 2000 Mar 1;164(5):2444-56.http://www.ncbi.nlm.nih.gov/pubmed/10679081?ordinalpos=29&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(193)
Strid J, Lopes L, Marcinkiewicz J,
Petrovska L, Nowak B, Chain BM, et al. A defect in bone marrow derived
dendritic cell maturation in the nonobesediabetic mouse. Clin Exp Immunol 2001
Mar;123(3):375-81.http://www.ncbi.nlm.nih.gov/pubmed/11298122?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(194)
Wong FS, Karttunen J, Dumont C, Wen L,
Visintin I, Pilip IM, et al. Identification of an MHC class I-restricted
autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat
Med 1999 Sep;5(9):1026-31.http://www.ncbi.nlm.nih.gov/pubmed/10470079?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(195)
Buchs AE, Rapoport MJ. T cell signaling
and autoimmune diabetes. J Pediatr Endocrinol Metab 2000 Nov;13(9):1549-54.http://www.ncbi.nlm.nih.gov/pubmed/11154149?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(196)
Zhang J, Salojin KV, Delovitch TL. CD28
co-stimulation restores T cell responsiveness in NOD mice by overcoming
deficiencies in Rac-1/p38 mitogen-activated protein kinase signaling and IL-2
and IL-4 gene transcription. Int Immunol 2001 Mar;13(3):377-84.http://www.ncbi.nlm.nih.gov/pubmed/11222507?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(197)
King C, Ilic A, Koelsch K, Sarvetnick N.
Homeostatic expansion of T cells during immune insufficiency generates
autoimmunity. Cell 2004 Apr 16;117(2):265-77.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15084263
(198)
Lang J, Bellgrau D. A T-cell functional
phenotype common among autoimmune-prone rodent strains. Scand J Immunol 2002
Jun;55(6):546-59.http://www.ncbi.nlm.nih.gov/pubmed/12028557?ordinalpos=21&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(199)
McInerney MF, Pek SB, Thomas DW.
Prevention of insulitis and diabetes onset by treatment with complete Freund's
adjuvant in NOD mice. diab 1991;40(6):715-25.http://www.ncbi.nlm.nih.gov/pubmed/2040388?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(200) Guytingco R, Quddus J, Richardson B, Pek SB.
Single administration of bacillus Calmette-Guerin (BCG) vaccine in nonobese
diabetic (NOD) mice results in suppression of in vitro T-lymphocyte
proliferation and regression of insulitis. Diabetes 40, 152A. 1991.
Ref Type: Abstract
(201)
Singh B. Stimulation of the developing
immune system can prevent autoimmunity. J Autoimmun 2000 Feb;14(1):15-22.http://www.ncbi.nlm.nih.gov/pubmed/10648113?ordinalpos=13&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(202)
Ridgway WM, Fassò M, Lanctot A, Garvey
C, Fathman CG. Breaking self-tolerance in nonobese diabetic mice. J Exp Med
1996;183:1657-62.http://www.ncbi.nlm.nih.gov/pubmed/8666923?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(203)
Boyton RJ, Lohmann T, Londei M,
Kalbacher H, Halder T, Frater AJ, et al. Glutamic acid decarboxylase T
lymphocyte responses associated with susceptibility or resistance to type I
diabetes: analysis in disease discordant human twins, non-obese diabetic mice
and HLA-DQ transgenic mice. Int Immunol 1998 Dec;10(12):1765-76.http://www.ncbi.nlm.nih.gov/pubmed/9885897?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(204)
Dosch HM, Becker DJ. Measurement of
T-cell autoreactivity in autoimmune diabetes [letter] [In Process Citation].
diabetol 2000 Mar;43(3):386-7.http://www.ncbi.nlm.nih.gov/pubmed/10768103?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(205)
Kreuwel HT, Biggs JA, Pilip IM, Pamer
EG, Lo D, Sherman LA. Defective CD8+ T cell peripheral tolerance in nonobese
diabetic mice. J Immunol 2001 Jul 15;167(2):1112-7.http://www.ncbi.nlm.nih.gov/pubmed/11441123?ordinalpos=11&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(206)
Tian J, Gregori S, Adorini L, Kaufman
DL. The frequency of high avidity T cells determines the hierarchy of
determinant spreading. J Immunol 2001 Jun 15;166(12):7144-50.http://www.ncbi.nlm.nih.gov/pubmed/11390460?ordinalpos=139&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(207)
Hutton JC, Eisenbarth GS. A pancreatic
{beta}-cell-specific homolog of glucose-6-phosphatase emerges as a major target
of cell-mediated autoimmunity in diabetes. Proc Natl Acad Sci U S A 2003 Jul
14;100:8626-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=12861077
(208)
Trudeau JD, Kelly-Smith C, Verchere CB,
Elliott JF, Dutz JP, Finegood DT, et al. Prediction of spontaneous autoimmune
diabetes in NOD mice by quantification of autoreactive T cells in peripheral
blood. J Clin Invest 2003 Jan;111(2):217-23.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12531877
(209)
Amrani A, Verdaguer J, Serra P, Tafuro
S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity
maturation of a T-cell population. Nature 2000 Aug 17;406(6797):739-42.http://www.ncbi.nlm.nih.gov/pubmed/10963600?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(210)
Naquet P, Naspetti M, Boyd R.
Development, organization and function of the thymic medulla in normal,
immunodeficient or autoimmune mice. Semin Immunol 1999 Feb;11(1):47-55.http://www.ncbi.nlm.nih.gov/pubmed/9950751?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(211)
Lord CJ, Howlett S, Lyons PA, Peterson
LB, Wicker LS, Todd JA. The murine type 1 diabetes loci, Idd1, Idd3, Idd5,
Idd9, and Idd17/10/18, do not control thymic CD4-CD8-/TCRalphabeta+ deficiency
in the nonobese diabetic mouse. Mamm Genome 2001 Feb;12(2):175-6.http://www.ncbi.nlm.nih.gov/pubmed/11210190?ordinalpos=25&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(212)
Bluestone JA, Tang Q. Therapeutic
vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc Natl Acad
Sci U S A 2004 Oct 5;101 Suppl 2:14622-6.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15322272
(213)
Tang Q, Bluestone JA. The Foxp3+
regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 2008
Mar;9(3):239-44.http://www.ncbi.nlm.nih.gov/pubmed/18285775?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(214)
Masteller EL, Warner MR, Tang Q, Tarbell
KV, McDevitt H, Bluestone JA. Expansion of functional endogenous
antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J
Immunol 2005 Sep 1;175(5):3053-9.http://www.ncbi.nlm.nih.gov/pubmed/16116193?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(215)
Hutchings PR, Cooke A. The transfer of
autoimmune diabetes in NOD mice can be inhibited or accelerated by distinct
cell populations present in normal splenocytes taken from young males. J
Autoimmun 1990 Apr;3(2):175-85.http://www.ncbi.nlm.nih.gov/pubmed/1971173?ordinalpos=9&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(216)
Boitard C, Yasunami R, Dardenne M, Bach
JF. T cell-mediated inhibition of the transfer of autoimmune diabetes in NOD
mice. J Exp Med 1989 May 1;169(5):1669-80.http://www.ncbi.nlm.nih.gov/pubmed/2523954?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(217)
Belghith M, Bluestone JA, Barriot S,
Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate
restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune
diabetes. Nat Med 2003 Sep;9(9):1202-8.http://www.ncbi.nlm.nih.gov/pubmed/12937416?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(218)
Weiner HL. Oral tolerance, an active
immunologic process mediated by multiple mechanisms. J Clin Invest 2000
Oct;106(8):935-7.http://www.ncbi.nlm.nih.gov/pubmed/11032852?ordinalpos=13&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(219)
Roncarolo MG, Gregori S. Is FOXP3 a bona
fide marker for human regulatory T cells? Eur J Immunol 2008 Apr;38(4):925-7.http://www.ncbi.nlm.nih.gov/pubmed/18395862?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(220)
Trucco M, Giannoukakis N.
Immunoregulatory dendritic cells to prevent and reverse new-onset Type 1
diabetes mellitus. Expert Opin Biol Ther 2007 Jul;7(7):951-63.http://www.ncbi.nlm.nih.gov/pubmed/17665986?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(221)
Du W, Wong FS, Li MO, Peng J, Qi H,
Flavell RA, et al. TGF-beta signaling is required for the function of
insulin-reactive T regulatory cells. J Clin Invest 2006 May 1;116(5):1360-70.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=16670772
(222)
Pop SM, Wong CP, He Q, Wang Y, Wallet
MA, Goudy KS, et al. The type and frequency of immunoregulatory CD4+ T-cells
govern the efficacy of antigen-specific immunotherapy in nonobese diabetic
mice. Diabetes 2007 May;56(5):1395-402.http://www.ncbi.nlm.nih.gov/pubmed/17317763?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(223)
Fehervari Z, Sakaguchi S. CD4(+) Tregs
and immune control. J Clin Invest 2004 Dec;114(9):1209-17.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15520849
(224)
Hori S, Nomura T, Sakaguchi S. Control
of regulatory T cell development by the transcription factor Foxp3. Science
2003 Feb 14;299(5609):1057-61.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12522256
(225)
Fontenot JD, Gavin MA, Rudensky AY.
Foxp3 programs the development and function of CD4+CD25+ regulatory T cells.
Nat Immunol 2003 Apr;4(4):330-6.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12612578
(226)
Jaeckel E, von Boehmer H, Manns MP.
Antigen-Specific FoxP3-Transduced T-Cells Can Control Established Type 1
Diabetes. diab 2005;54(2):306-10.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15591438
(227)
Mchugh RS, Shevach EM. Cutting edge:
Depletion of CD4(+)CD25(+) regulatory T cells is necessary, but not sufficient,
for induction of organ-specific autoimmune disease. Journal of Immunology
2002;168(12):5979-83.http://www.ncbi.nlm.nih.gov/pubmed/12055202?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(228)
Peng Y, Laouar Y, Li MO, Green EA,
Flavell RA. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+
regulatory T cells responsible for protection against diabetes. Proc Natl Acad
Sci U S A 2004 Mar 30;101(13):4572-7.http://www.ncbi.nlm.nih.gov/pubmed/15070759?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(229)
Tang Q, Henriksen KJ, Bi M, Finger EB,
Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells
suppress autoimmune diabetes. J Exp Med 2004 Jul 7;199(11):1455-65.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15184499
(230)
Lenschow DJ, Herold KC, Rhee L, Patel B,
Koons A, Qin HY, et al. CD28/B7 regulation of Th1 and Th2 subsets in the
development of autoimmune diabetes [published erratum appears in Immunity 1997
Feb;6(2):following 215]. Immunity 1996 Sep;5(3):285-93.http://www.ncbi.nlm.nih.gov/pubmed/8808683?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(231)
Herman AE, Freeman GJ, Mathis D, Benoist
C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of
effector cells in the prediabetic lesion. J Exp Med 2004 Jun 7;199(11):1479-89.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15184501
(232)
Mukherjee R, Chaturvedi P, Qin HY, Singh
B. CD4+CD25+ regulatory T cells generated in response to insulin B:9-23 peptide
prevent adoptive transfer of diabetes by diabetogenic T cells. J Autoimmun 2003
Nov;21(3):221-37.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14599847
(233)
Harrison LC, Solly NR, Martinez NR.
(Pro)insulin-specific regulatory T cells. Novartis Found Symp 2003;252:132-41.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14609216
(234)
Gombert JM, Herbelin A, Tancrede-Bohin
E, Dy M, Carnaud C, Bach JF. Early quantitative and functional deficiency of
NK1+-like thymocytes in the NOD mouse. Eur J Immunol 1996;26(12):2989-98.http://www.ncbi.nlm.nih.gov/pubmed/8977295?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(235)
Falcone M, Yeung B, Tucker L, Rodriguez
E, Sarvetnick N. A defect in interleukin 12-induced activation and interferon
gamma secretion of peripheral natural killer T cells in nonobese diabetic mice
suggests new pathogenic mechanisms for insulin-dependent diabetes mellitus. J
Exp Med 1999 Oct 4;190(7):963-72.http://www.ncbi.nlm.nih.gov/pubmed/10510086?ordinalpos=5&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(236)
Lee IF, Qin H, Trudeau J, Dutz J, Tan R.
Regulation of autoimmune diabetes by complete Freund's adjuvant is mediated by
NK cells. J Immunol 2004 Jan 15;172(2):937-42.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14707066
(237)
Homann D, Jahreis A, Wolfe T, Hughes A,
Coon B, van Stipdonk MJ, et al. CD40L blockade prevents autoimmune diabetes by
induction of bitypic NK/DC regulatory cells. Immunity 2002 Mar;16(3):403-15.http://www.ncbi.nlm.nih.gov/pubmed/11911825?ordinalpos=9&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(238)
Wegmann DR, Norbury-Glaser M, Daniel D.
Insulin-specific T cells are a predominant component of islet infiltrates in
pre-diabetic NOD mice. Eur J Immunol 1994;24(8):1853-7.http://www.ncbi.nlm.nih.gov/pubmed/8056042?ordinalpos=7&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(239)
Lieberman SM, Evans AM, Han B, Takaki T,
Vinnitskaya Y, Caldwell JA, et al. Identification of the {beta} cell antigen
targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune
diabetes. Proc Natl Acad Sci U S A 2003 Jul 8;100(14):8384-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12815107
(240)
Lieberman SM, DiLorenzo TP. A
comprehensive guide to antibody and T-cell responses in type 1 diabetes. Tissue
Antigens 2003 Nov;62(5):359-77.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14617043
(241)
Li SW, Koya V, Li Y, Donelan W, Lin P,
Reeves WH, et al. Pancreatic duodenal homeobox 1 protein is a novel
beta-cell-specific autoantigen for type I diabetes. Lab Invest 2010
Jan;90(1):31-9.PM:19901909
(242)
Gurr W, Shaw M, Li Y, Sherwin R. RegII
is a beta-cell protein and autoantigen in diabetes of NOD mice. Diabetes 2007
Jan;56(1):34-40
(243)
Jaeckel E, Klein L, Martin-Orozco N, von
Boehmer H. Normal incidence of diabetes in NOD mice tolerant to glutamic acid
decarboxylase. J Exp Med 2003 Jun 16;197(12):1635-44.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12796471
(244)
Lieberman SM, Takaki T, Han B,
Santamaria P, Serreze DV, DiLorenzo TP. Individual nonobese diabetic mice
exhibit unique patterns of CD8+ T cell reactivity to three islet antigens,
including the newly identified widely expressed dystrophia myotonica kinase. J
Immunol 2004 Dec 1;173(11):6727-34.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15557165
(245)
Cohen IR. Thoughts on the pathogenesis
of type 1 diabetes and on the arrest of autoimmune beta-cell destruction by
peptide p277 vaccination. Isr Med Assoc J 2004 May;6(5):260-1.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15151361
(246)
Bonifacio E, Atkinson M, Eisenbarth GS,
Serreze D, Kay TW, Lee-Chan E, et al. International workshop on autoimmunity in
animal models of autoimmune diabetes identifies insulin, but not GAD or IA-2 as
specific antigens of humoral autoimmunity in the non-obese diabetic mouse. diab
2001;50:2451-8.http://www.ncbi.nlm.nih.gov/pubmed/11679421?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(247)
Daniel D, Gill RG, Schloot N, Wegmann D.
Epitope specificity, cytokine production profile and diabetogenic activity of
insulin-specific T cell clones isolated from NOD mice. Eur J Immunol
1995;25(4):1056-62.http://www.ncbi.nlm.nih.gov/pubmed/7537670?ordinalpos=15&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(248)
Jaeckel E, Lipes MA, von Boehmer H.
Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1
diabetes. Nat Immunol 2004 Oct;5(10):1028-35.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15378058
(249)
Narendran P, Mannering SI, Harrison LC.
Proinsulin - a pathogenic autoantigen in type 1 diabetes. Autoimmunity Reviews
2003;2(4):204-10.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12848947
(250) Muir A, Luchetta R, Song H-Y, Peck A, Krischer
J, Maclaren N. Insulin immunization protects NOD mice from diabetes.
Autoimmunity 15, 58. 1993.
Ref Type: Abstract
(251)
Muir A, Peck A, Clare-Salzler M, Song
Y-H, Cornelius J, Luchetta R, et al. Insulin immunization of nonobese diabetic
mice induces a protective insulitis characterized by diminished intraislet
interferon-gamma transcription. J Clin Invest 1995;95(2):628-34.http://www.ncbi.nlm.nih.gov/pubmed/7860747?ordinalpos=25&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(252)
Hancock WW, Polansky M, Zhang J, Blogg
N, Weiner HL. Suppression of insulitis in non-obese diabetic (NOD) mice by oral
insulin administration is associated with selective expression of interleukin-4
and -10, transforming growth factor-beta, and prostaglandin-E. Am J Pathol 1995;147(5):1193-9.http://www.ncbi.nlm.nih.gov/pubmed/7485382?ordinalpos=30&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(253)
Zhang ZJ, Davidson L, Eisenbarth G,
Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral
administration of porcine insulin. Proc Natl Acad Sci U S A 1991 Nov
15;88(22):10252-6.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=1946445
(254)
Simone E, Daniel D, Schloot N, Gottlieb
P, Babu S, Kawasaki E, et al. T cell receptor restriction of diabetogenic
autoimmune NOD T cells. Proc Natl Acad Sci USA 1997;94(6):2518-21
(255)
Simone EA, Yu L, Wegmann DR, Eisenbarth
GS. T cell receptor gene polymorphisms associated with anti-insulin, autoimmune
T cells in diabetes-prone NOD mice. J Autoimmun 1997;10:317-21
(256)
Abiru N, Wegmann D, Kawasaki E, Gottlieb
P, Simone E, Eisenbarth GS. Dual overlapping peptides recognized by insulin
peptide B:9-23 reactive T cell receptor AV13S3 T cell clones of the NOD mouse.
J Autoimmun 2000;14(3):231-7
(257)
Daniel D, Wegmann DR. Protection of
nonobese diabetic mice from diabetes by intranasal or subcutaneous
administration of insulin peptide B-(9-23). Proc Natl Acad Sci USA
1996;93(2):956-60.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=8570667&query_hl=58&itool=pubmed_docsum
(258)
Wong FS, Janeway CAJ. The role of CD4
vs. CD8 T cells in IDDM. J Autoimmun 1999 Nov;13(3):290-5
(259)
Tsai S, Shameli A, Yamanouchi J,
Clemente-Casares X, Wang J, Serra P, et al. Reversal of autoimmunity by
boosting memory-like autoregulatory T cells. Immunity 2010 Apr
23;32(4):568-80.PM:20381385
(260)
Kash SF, Condie BG, Baekkeskov S.
Glutamate decarboxylase and GABA in pancreatic islets: lessons from knock-out
mice. Horm Metab Res 1999 May;31(5):340-4
(261)
Kubosaki A, Gross S, Miura J, Saeki K,
Zhu M, Nakamura S, et al. Targeted disruption of the IA-2beta gene causes
glucose intolerance and impairs insulin secretion but does not prevent the
development of diabetes in NOD mice. diab 2004 Jul;53(7):1684-91.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15220191
(262)
Kubosaki A, Miura J, Notkins AL. IA-2 is
not required for the development of diabetes in NOD mice. diabetol 2004
Jan;47(1):149-50.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14614561
(263)
Kim SK, Tarbell KV, Sanna M,
Vadeboncoeur M, Warganich T, Lee M, et al. Prevention of type I diabetes
transfer by glutamic acid decarboxylase 65 peptide 206-220-specific T cells.
Proc Natl Acad Sci U S A 2004 Sep 28;101(39):14204-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15381770
(264)
Burton AR, Vincent E, Arnold PY, Lennon
GP, Smeltzer M, Li CS, et al. On the pathogenicity of autoantigen-specific
T-cell receptors. Diabetes 2008 May;57(5):1321-30.http://www.ncbi.nlm.nih.gov/pubmed/18299317?ordinalpos=7&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(265)
Burton AR, Baquet Z, Eisenbarth GS,
Tisch R, Smeyne R, Workman CJ, et al. Central Nervous System Destruction
Mediated by Glutamic Acid Decarboxylase-Specific CD4+ T Cells. J Immunol 2010
Mar 26.PM:20348424
(266)
Zekzer D, Wong FS, Ayalon O, Millet I,
Altieri M, Shintani S, et al. GAD-reactive CD4+ Th1 cells induce diabetes in
NOD/SCID mice. J Clin Invest 1998 Jan 1;101(1):68-73.PM:9421467
(267)
Yoon JW, Yoon CS, Lim HW, Huang QQ, Kang
Y, Pyun KH, et al. Control of autoimmune diabetes in NOD mice by GAD expression
or suppression in beta cells. Science 1999 May 14;284(5417):1183-7
(268)
Gebe JA, Unrath KA, Yue BB, Miyake T,
Falk BA, Nepom GT. Autoreactive human T-cell receptor initiates insulitis and
impaired glucose tolerance in HLA DR4 transgenic mice. J Autoimmun 2008
Jun;30(4):197-206.http://www.ncbi.nlm.nih.gov/pubmed/17949947?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(269)
Quintana FJ, Carmi P, Cohen IR. DNA
vaccination with heat shock protein 60 inhibits cyclophosphamide-accelerated
diabetes. J Immunol 2002 Nov 15;169(10):6030-5.PM:12421990
(270)
Ablamunits V, Elias D, Reshef T, Cohen
IR. Islet T cells secreting IFN-gamma in NOD mouse diabetes: arrest by p277
peptide treatment. J Autoimmun 1998 Feb;11(1):73-81
(271)
Elias D, Meilin A, Ablamunits V, Birk
OS, Carmi P, Könen-Waisman S, et al. Hsp60 peptide therapy of NOD mouse
diabetes induces a Th2 cytokine burst and downregulates autoimmunity to various
b-cell antigens.
diab 1997;46:758-64
(272)
Knip M, Karjalainen J, Akerblom HK.
Islet cell antibodies are less predictive of IDDM among unaffected children in
the general population than in sibs of children with diabetes. The Childhood
Diabetes in Finland Study Group. Diab care 1998;21(10):1670-3
(273)
Bowman M, Atkinson MA. Heat shock
protein therapy fails to prevent diabetes in NOD mice. diabetol 2002
Sep;45(9):1350-1.PM:12242470
(274)
Faideau B, Briand JP, Lotton C, Tardivel
I, Halbout P, Jami J, et al. Expression of preproinsulin-2 gene shapes the
immune response to preproinsulin in normal mice. J Immunol 2004 Jan
1;172(1):25-33.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14688305
(275)
Devendra D, Paronen J, Moriyama H, Miao
D, Eisenbarth GS, Liu E. Differential immune response to B:9-23 insulin 1 and
insulin 2 peptides in animal models of type 1 diabetes. Journal of Autoimmunity
2004;23(1):17-26.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15236749
(276)
French MB, Allison J, Cram DS, Thomas
HE, Dempsey-Collier M, Silva A, et al. Transgenic expression of mouse
proinsulin II prevents diabetes in nonobese diabetic mice. diab 1996;46:34-9
(277)
Steptoe RJ, Ritchie JM, Harrison LC.
Transfer of hematopoietic stem cells encoding autoantigen prevents autoimmune
diabetes. J Clin Invest 2003 May;111(9):1357-63.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12727927
(278)
DiLorenzo TP, Serreze DV. The good
turned ugly: immunopathogenic basis for diabetogenic CD8+ T cells in NOD mice.
Immunol Rev 2005 Apr;204:250-63.:250-63
(279)
Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar
SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major
autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007 Oct
23;104(43):17040-5.http://www.ncbi.nlm.nih.gov/pubmed/17942684?ordinalpos=8&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(280)
Krishnamurthy B, Dudek NL, McKenzie MD,
Purcell AW, Brooks AG, Gellert S, et al. Responses against islet antigens in
NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest
2006 Dec 1;116(12):3258-65.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=17143333&query_hl=7&itool=pubmed_docsum
(281)
Nakhooda AF, Like AA, Chappel CI, Murray
FT, Marliss EB. The spontaneously diabetic Wistar rat: metabolic and
morphologic studies. diab 2001;26:100-12
(282)
Like AA, Rossini AA. Spontaneous autoimmune
diabetes mellitus in the BioBreeding/Worcester rat. Surv Synth Path Res
1984;3(2):131-8
(283)
Colle E, Guttmann RD, Seemayer T.
Spontaneous diabetes mellitus syndrome in the rat. I. Association with the
major histocompatibility complex. J Exp Med 1981;154(4):1237-42
(284)
Guttmann RD, Colle E, Michel F, Seemayer
T. Spontaneous diabetes mellitus syndrome in the rat. II. T lymphopenia and its association with
clinical disease and pancreatic lymphocytic infiltration. J Immunol 1983;130(4):1732-5
(285)
Elder ME, Maclaren NK. Identification of
profound peripheral T lymphocyte immunodeficiencies in the spontaneously
diabetic BB rat. J Immunol 1983 Apr;130(4):1723-31.PM:6220066
(286)
Bellgrau D, Naji A, Silvers WK, Markmann
JF, Barker CF. Spontaneous diabetes in BB rats: evidence for T cell dependent
immune response defect. diabetol 1982;23(4):359-64
(287)
Woda BA, Like AA, Padden C, McFadden ML.
Deficiency of phenotypic cytotoxic-suppressor T lymphocytes in the BB/W rat. J
Immunol 1986 Feb 1;136(3):856-9.PM:2934480
(288)
Sellins KS, Gold DP, Bellgrau D.
Resistance to tolerance induction in the diabetes-prone biobreeding rat as one
manifestation of abnormal responses to superantigens. diabetol 1996;39(1):28-36
(289)
Crisa L, Mordes JP, Rossini AA.
Autoimmune diabetes mellitus in the BB rat. Diabetes Metab Rev 1992;8(1):4-37
(290)
Mordes JP, Bortell R, Doukas J, Rigby M,
Whalen B, Zipris D, et al. The BB/Wor rat and the balance hypothesis of
autoimmunity. Diabetes Metab Rev 1996 Jul;12(2):103-9
(291)
Prins JB, Herberg L, den Bieman M, van
Zutpen B. Genetic variation within and between lines of diabetes-prone and
non-diabetes-prone BB rats: allele distribution of 8 protein markers. Lab Anim
2001;25:207-11
(292)
Butler L, Guberski DL, Like A. Changes
in penetrance and onset of sontaneous diabetes in the BB/Wor rat. In: Shafrir
E, editor. Frontier in Diabetes Research II. Lessons from Animal Diabetes III.
1990. p. 50-3.
(293)
Zipris D, Lien E, Nair A, Xie JX,
Greiner DL, Mordes JP, et al. TLR9-signaling pathways are involved in Kilham
rat virus-induced autoimmune diabetes in the biobreeding diabetes-resistant
rat. J Immunol 2007 Jan 15;178(2):693-701.http://www.ncbi.nlm.nih.gov/sites/entrez?orig_db=PubMed&db=PubMed&cmd=Search&term=178%5Bvolume%5D%20AND%20693%5Bpage%5D%20AND%202007%5Bpdat%5D
(294)
Colle E, Fuks A, Poussier P, Edouard P,
Guttman RD. Polygenic nature of spontaneous diabetes in the rat: Permisive MHC haplotype and presence of the
lymphopenic trait of the BB rat are not sufficient to produce susceptibility.
diab 1992;41:1617-23
(295)
Jackson RA, Buse JB, Rifai R, Pelletier
D, Milford EL, Carpenter CB, et al. Two genes required for diabetes in BB rats.
Evidence from cyclical intercrosses and backcrosses. J Exp Med
1984;159(6):1629-36
(296)
Pandarpurkar M, Wilson-Fritch L, Corvera
S, Markholst H, Hornum L, Greiner DL, et al. Ian4 is required for mitochondrial
integrity and T cell survival. Proc Natl Acad Sci U S A 2003 Sep
2;100(18):10382-7.http://www.ncbi.nlm.nih.gov/pubmed/12930893?ordinalpos=6&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(297)
Lang JA, Kominski D, Bellgrau D,
Scheinman RI. Partial activation precedes apoptotic death in T cells harboring
an IAN gene mutation. Eur J Immunol 2004 Sep;34(9):2396-406.http://www.ncbi.nlm.nih.gov/pubmed/15307172?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(298)
Nitta T, Nasreen M, Seike T, Goji A,
Ohigashi I, Miyazaki T, et al. IAN family critically regulates survival and
development of T lymphocytes. PLoS Biol 2006 Apr;4(4):e103.http://www.ncbi.nlm.nih.gov/pubmed/16509771?ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(299)
Ellerman KE, Like AA. Susceptibility to
diabetes is widely distributed in normal class IIu haplotype rats. diabetol
2000 Jul;43(7):890-8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=10952462&query_hl=25&itool=pubmed_docsum
(300)
Penhale WJ, Stumbles PA, Huxtable CR,
Sutherland RJ, Pethick DW. Induction of diabetes in PVG/c strain rats by
manipulation of the immune system. Autoimmunity 1990;7(2-3):169-79.PM:2104183
(301)
Martin AM, Maxson MN, Leif J, Mordes JP,
Greiner DL, Blankenhorn EP. Diabetes-prone and diabetes-resistant BB rats share
a common major diabetes susceptibility locus, iddm4: additional evidence for a
"universal autoimmunity locus" on rat chromosome 4. diab 1999
Nov;48(11):2138-44.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=10535446
(302)
Kloting I, I, van den BJ, Kovacs P.
Quantitative trait loci for blood glucose confirm diabetes predisposing and
protective genes, Iddm4 and Iddm5r, in the spontaneously diabetic BB/OK rat.
Int J Mol Med 1998 Nov;2(5):597-601.PM:9858659
(303)
Wallis RH, Wang K, Dabrowski D, Marandi
L, Ning T, Hsieh E, et al. A novel susceptibility locus on rat chromosome 8
affects spontaneous but not experimentally induced type 1 diabetes. Diabetes
2007 Jun;56(6):1731-6.http://www.ncbi.nlm.nih.gov/pubmed/17389329?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(304)
Hornum L, Romer J, Markholst H. The
diabetes-prone BB rat carries a frameshift mutation in Ian4, a positional
candidate of Iddm1. diab 2002 Jun;51(6):1972-9.PM:12031988
(305)
MacMurray AJ, Moralejo DH, Kwitek AE,
Rutledge EA, Van Yserloo B, Gohlke P, et al. Lymphopenia in the BB Rat Model of
Type 1 Diabetes is Due to a Mutation in a Novel Immune-Associated Nucleotide
(Ian)-Related Gene. Genome Res 2002 Jul;12(7):1029-39.PM:12097339
(306)
Hernandez-Hoyos G, Joseph S, Miller NG,
Butcher GW. The lymphopenia mutation of the BB rat causes inappropriate
apoptosis of mature thymocytes. Eur J Immunol 1999 Jun;29(6):1832-41.PM:10382745
(307)
Blankenhorn EP, Cort L, Greiner DL,
Guberski DL, Mordes JP. Virus-induced autoimmune diabetes in the LEW.1WR1 rat
requires Iddm14 and a genetic locus proximal to the major histocompatibility
complex. diab 2009 Dec;58(12):2930-8.PM:19720792
(308)
Guberski DL, Thomas VA, Shek WR, Like
AA, Handler ES, Rossini AA, et al. Induction of Type 1 diabetes by Kilham's rat
virus in diabetes-resistant BB/Wor rats. Science 1991;254:1010-3.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=1658938
(309)
Sobel DO, Newsome J, Ewel CH, Bellanti
JA, Abbassi V, Creswell K, et al. Poly I:C induces development of diabetes
mellitus in BB rat. diab 1992;41(4):515-20
(310)
Like AA. Depletion of RT6.1+ lymphocytes
alone is insufficient to induce diabetes in diabetes-resistant BB/WOR rats. Am
J Pathol 1990;136:565-74
(311)
Ramanathan S, Bihoreau MT, Paterson AD,
Marandi L, Gauguier D, Poussier P. Thymectomy and radiation-induced type 1
diabetes in nonlymphopenic BB rats. diab 2002 Oct;51(10):2975-81.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12351436
(312)
Ellerman KE, Richards CA, Guberski DL,
Shek WR, Like AA. Kilham rat virus triggers T-cell-dependent autoimmune
diabetes in multiple strains of rat. diab 1996;45:557-62
(313)
Chung YH, Jun HS, Son M, Bao M, Bae HY,
Kang Y, et al. Cellular and molecular mechanism for Kilham rat virus-induced
autoimmune diabetes in DR-BB rats. J Immunol 2000 Sep
1;165(5):2866-76.PM:10946320
(314)
Zipris D, Lien E, Xie JX, Greiner DL,
Mordes JP, Rossini AA. TLR activation synergizes with Kilham rat virus
infection to induce diabetes in BBDR rats. J Immunol 2005 Jan 1;174(1):131-42.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15611235
(315)
Hornum L, Lundsgaard D, Markholst H.
PolyI:C induction of diabetes is controlled by Iddm4 in rats with a full
regulatory T cell pool. Ann N Y Acad Sci 2007 Sep;1110:65-72.:65-72
(316)
Blankenhorn EP, DeScipio C, Rodemich L,
Cort L, Leif JH, Greiner DL, et al. Refinement of the Iddm4 diabetes
susceptibility locus reveals TCRVbeta4 as a candidate gene. Ann N Y Acad Sci
2007 Apr;1103:128-31. Epub@2007 Mar 21.:128-31
(317)
Hornum L, DeScipio C, Markholst H,
Troutman SA, Novak S, Leif J, et al. Comparative mapping of rat Iddm4 to
segments on HSA7 and MMU6. Mamm Genome 2004 Jan;15(1):53-61.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14727142
(318)
Kloting I, Schmidt S, Kovacs P. Mapping
of novel genes predisposing or protecting diabetes development in the BB/OK
rat. Biochem Biophys Res Commun 1998 Apr 17;245(2):483-6.PM:9571179
(319)
Jackerott M, Hornum L, Andreasen BE,
Markholst H. Segregation of autoimmune type 1 diabetes in a cross between
diabetic BB and brown Norway rats. J Autoimmun 1997 Feb;10(1):35-41.PM:9080298
(320)
Mordes JP, Cort L, Norowski E, Leif J,
Fuller JM, Lernmark A, et al. Analysis of the rat Iddm14 diabetes
susceptibility locus in multiple rat strains: identification of a
susceptibility haplotype in the Tcrb-V locus. Mamm Genome 2009
Mar;20(3):162-9.PM:19205800
(321)
Jackson R, Rassi N, Crump A, Haynes BF,
Eisenbarth GS. The BB diabetic rat. Profound T-cell lymphocytopenia. diab
1981;30(10):887-9
(322)
Ely JM, Greiner DL, Lubaroff DM, Fitch
FW. Characterization of monoclonal antibodies that define rat T cell
alloantigens. J Immunol 1983;130:2798-803
(323)
Greiner DL, Handler ES, Nakano K, Mordes
JP, Rossini AA. Absence of the RT-6 T cell subset in diabetes-prone BB/W rats.
J Immunol 1986;136:148-51
(324)
Groen H, Van Der Berk J, Nieuwenhuis P,
Kampinga J. Peripheral T cells in diabetes prone (DP) BB rats are
CD45R-negative. Thymus 1989;14:145-50
(325)
Spickett GP, Brandon MR, Mason DW,
Williams AF, Woollett GR. MRC OX-22, a monoclonal antibody that labels a new
subset of T lymphocytes and reacts with the high molecular weight form of the
leukocyte-common antigen. J Exp Med 1983 Sep 1;158(3):795-810.PM:6224884
(326)
Thomas VA, Woda BA, Handler ES, Greiner
DL, Mordes JP, Rossini AA. Altered expression of diabetes in BB/Wor rats by
exposure to viral pathogens. diab 1991;40(2):255-8
(327)
Whalen BJ, Greiner DL, Mordes JP,
Rossini AA. Adoptive transfer of autoimmune diabetes mellitus to athymic rats:
synergy of CD4+ and CD8+ T cells and prevention by RT6+ T cells. J Autoimmun
1994 Dec;7(6):819-31.PM:7888038
(328)
Rossini AA, Mordes JP, Greiner DL,
Nakano K, Appel MC, Handler ES. Spleen cell transfusion in the
bio-breeding/worcester rat. J Clin Invest 1986;77:1399-401
(329)
Hillebrands JL, Whalen B, Visser JT,
Koning J, Bishop KD, Leif J, et al. A regulatory CD4+ T cell subset in the BB
rat model of autoimmune diabetes expresses neither CD25 nor Foxp3. J Immunol
2006 Dec 1;177(11):7820-32
(330)
Holm TL, Lundsgaard D, Markholst H.
Characteristics of rat CD4(+)CD25(+) T cells and their ability to prevent not
only diabetes but also insulitis in an adoptive transfer model in BB rats.
Scand J Immunol 2006 Jul;64(1):17-29
(331)
Georgiou HM, Lagarde AC, Bellgrau D. T
cell dysfunction in the diabetes-prone BB rat: a role for thymic migrants that
are not T cell precursors. J Exp Med 1988;167:132-48
(332)
Bellgrau D, Redd JM, Sellins KS.
Peculiar T-cell signaling does not preclude positive selection in the diabetes-prone
BB rat. diab 1994;43(1):47-52
(333)
Hosszufalusi N, Chan E, Granger G,
Charles MA. Quantitative analyses comparing all major spleen cell phenotypes in
BB and normal rats: autoimmune imbalance and double negative T cells associated
with resistant, prone and diabetic animals. J Autoimmun 1992
Jun;5(3):305-18.PM:1388637
(334)
Moore JK, Bellgrau D. Promiscuous
activation and cell cycle entry in T cells from autoimmune animals. Transplant
Proc 1999 May;31(3):1606-10.PM:10331020
(335)
Bieg S, Möller C, Olsson T, Lernmark Å.
The lymphopenia (lyp) gene controls
the intrathymic cytokine ratio in congenic BioBreeding rats. diabetol
1997;40:786-92
(336)
Bieg S. Differential expression of
p95vav in primary lymphoid tissue of BB rats congenic for the lymphopenia gene.
Autoimmunity 1999;30(1):37-42.PM:10433093
(337)
Zadeh HH, Greiner DL, Wu DY, Tausche F,
Goldschneider I. Abnormalities in the export and fate of recent thymic
emigrants in diabetes-prone BB/W rats. Autoimmunity 1996;24(1):35-46.PM:8937686
(338)
Moore JK, Scheinman RI, Bellgrau D. The
identification of a novel T cell activation state controlled by a diabetogenic
gene. J Immunol 2001 Jan 1;166(1):241-8.PM:11123298
(339)
Ramanathan S, Norwich K, Poussier P.
Antigen activation rescues recent thymic emigrants from programmed cell death
in the BB rat. J Immunol 1998 Jun 15;160(12):5757-64.PM:9637485
(340)
Delemarre FG, Simons PJ, de Heer HJ,
Drexhage HA. Signs of immaturity of splenic dendritic cells from the autoimmune
prone biobreeding rat: consequences for the in vitro expansion of regulator and
effector T cells. J Immunol 1999 Feb 1;162(3):1795-801.PM:9973444
(341)
Delemarre FG, Hoogeveen PG, Haan-Meulman
M, Simons PJ, Drexhage HA. Homotypic cluster formation of dendritic cells, a close
correlate of their state of maturation. Defects in the biobreeding
diabetes-prone rat. J Leukoc Biol 2001 Mar;69(3):373-80.PM:11261783
(342)
Iwakoshi NN, Greiner DL, Rossini AA,
Mordes JP. Diabetes prone BB rats are severely deficient in natural killer T
cells. Autoimmunity 1999;31(1):1-14.PM:10593564
(343)
Tullin S, Farris P, Petersen JS, Hornum
L, Jackerott M, Markholst H. A pronounced thymic B cell deficiency in the
spontaneously diabetic BB rat. J Immunol 1997 Jun 1;158(11):5554-9.PM:9164980
(344)
Rozing J, Coolen C, Tielen FJ, Weegenaar
J, Schuurman HJ, Greiner DL, et al. Defects in the thymic epithelial stroma of
diabetes prone BB rats. Thymus 1989;14(1-3):125-35.PM:2623736
(345)
Bellgrau D, Lagarde A-C. Cytotoxic
T-cell precursors with low-level CD8 in the diabetes-prone biobreeding rat:
implications for the generation of an autoimmune T-cell repertoire. Proc Natl
Acad Sci USA 1990;87:313-7
(346)
Groen H, Klatter FA, Brons NH, Mesander
G, Nieuwenhuis P, Kampinga J. Abnormal thymocyte subset distribution and
differential reduction of CD4+ and CD8+ T cell subsets during peripheral
maturation in diabetes-prone BioBreeding rats. J Immunol 1996 Feb
1;156(3):1269-75.PM:8558007
(347)
Plamondon C, Kottis V, Brideau C,
Metroz-Dayer MD, Poussier P. Abnormal thymocyte maturation in spontaneously
diabetic BB rats involves the deletion of CD4-8+ cells. J Immunol 1990 Feb
1;144(3):923-8.PM:1688592
(348)
Whalen BJ, Weiser P, Marounek J, Rossini
AA, Mordes JP, Greiner DL. Recapitulation of normal and abnormal BioBreeding
rat T cell development in adult thymus organ culture. J Immunol 1999 Apr
1;162(7):4003-12.PM:10201921
(349)
McKeever U, Mordes JP, Greiner DL, Appel
MC, Rozing J, Handler ES, et al. Adoptive transfer of autoimmune diabetes and
thyroiditis to athymic rats. Proc Natl Acad Sci U S A 1990
Oct;87(19):7618-22.PM:2217193
(350)
Sarkar P, Crisa L, McKeever U, Bortell
R, Handler E, Mordes JP, et al. Loss of RT6 message and most circulating T
cells after thymectomy of diabetes prone BB rats. Autoimmunity
1994;18(1):15-22.PM:7999952
(351)
Jung CG, Kamiyama T, Agui T. Elevated
apoptosis of peripheral T lymphocytes in diabetic BB rats. Immunology 1999
Dec;98(4):590-4.PM:10594693
(352)
Crispe IN, Dao T, Klugewitz K, Mehal WZ,
Metz DP. The liver as a site of T-cell apoptosis: graveyard, or killing field?
Immunol Rev 2000 Apr;174:47-62.PM:10807506
(353)
Scott J. The spontaneously diabetic BB
rat: sites of the defects leading to autoimmunity and diabetes mellitus. A
review. Curr Top Microbiol Immunol 1990;156:1-14.PM:2199161
(354)
Francfort JW, Naji A, Silvers WK, Barker
CF. The influence of T-lymphocyte precursor cells and thymus grafts on the
cellular immunodeficiencies of the BB rat. diab 1985;34:1134-8
(355)
Like A, Rossini AA, Guberski DL, Appel
MC. Spontaneous diabetes mellitus: Reversal and prevention in the BB/W rat with
antiserum to rat lymphocytes. Science 1979;206:1421-3
(356)
Like AA, Biron CA, Weringer EJ, Byman K,
Sroczynski E, Guberski DL. Prevention of diabetes in BioBreeding/Worcester rats
with monoclonal antibodies that recognize T lymphocytes or natural killer
cells. J Exp Med 1986;164(4):1145-59
(357)
Barlow AK, Like AA. Anti-CD2 monoclonal
antibodies prevent spontaneous and adoptive transfer of diabetes in the BB/Wor
rat. Am J Pathol 1992 Nov;141(5):1043-51.PM:1359788
(358)
Like AA, Kislauskis E, Williams RR,
Rossini AA. Neonatal thymectomy prevents spontaneous diabetes mellitus in the
BB/W rat. Science 1982;216(4546):644-6
(359)
Murase N, Lieberman I, Nalesnik M, Mintz
D, Todo S, Drash AL, et al. Prevention of spontaneous diabetes in BB rats with
FK 506. Lancet 1990 Aug 11;336(8711):373-4.PM:1697397
(360)
Nicoletti F, Meroni PL, Barcellini W,
Grasso S, Borghi MO, Lunetta M, et al. FK-506 prevents diabetes in
diabetes-prone BB/Wor rats. Int J Immunopharmacol 1991;13(7):1027-30.PM:1722192
(361)
Sadelain MW, Qin HY, Sumoski W, Parfrey
N, Singh B, Rabinovitch A. Prevention of diabetes in the BB rat by early
immunotherapy using Freund's adjuvant. J Autoimmun 1990 Dec;3(6):671-80
(362)
Like AA, Guberski DL, Butler L.
Influence of environmental viral agents on frequency and tempo of diabetes
mellitus in BB/Wor rats. diab 1991;40:259-62
(363)
Dryberg T, Schwimmbeck PL, Oldstone MBA.
Inhibition of diabetes in BB rats by virus infection. J Clin Invest
1988;81:928-31
(364)
Greiner DL, Malkani S, Kanaitsuka T,
Bortell R, Doukas J, Rigby M, et al. The T cell marker RT6 in a rat model of
autoimmune diabetes. Adv Exp Med Biol 1997;419:209-16.PM:9193656
(365)
Rossini AA, Faustman D, Woda BA, Like
AA, Szymanski I, Mordes JP. Lymphocyte transfusions prevent diabetes in the
Bio-Breeding/ Worcester rat. J Clin Invest 1984;74(1):39-46
(366)
Burstein D, Mordes JP, Greiner DL, Stein
D, Nakamura N, Handler ES, et al. Prevention of diabetes in BB/Wor rat by
single transfusion of spleen cells. Parameters that affect degree of
protection. diab 1989 Jan;38(1):24-30.PM:2642432
(367)
Ramanathan S, Poussier P. T cell
reconstitution of BB/W rats after the initiation of insulitis precipitates the
onset of diabetes. J Immunol 1999 May 1;162(9):5134-42.PM:10227984
(368)
Satoh J, Seino H, Shintani S, Tanaka S,
Ohteki T, Masuda T, et al. Inhibition of type 1 diabetes in BB rats with
recombinant human tumor necrosis factor-alpha. J Immunol 1990 Sep
1;145(5):1395-9.PM:2384663
(369)
Whalen BJ, Marounek J, Weiser P, Appel
MC, Greiner DL, Mordes JP, et al. BB rat thymocytes cultured in the presence of
islets lose their ability to transfer autoimmune diabetes. diab 2001 May;50(5):972-9.http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11334440
(370)
Brugman S, Klatter FA, Visser JT,
Wildeboer-Veloo AC, Harmsen HJ, Rozing J, et al. Antibiotic treatment partially
protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the
gut flora involved in the development of type 1 diabetes? Diabetologia 2006
Sep;49(9):2105-8
(371)
Kawano K, Hirashima T, Moris S, Saitoh
Y, Kurosumi M, Natori T. New inbred strain of Long-Evans Tokushima lean rats
with IDDM without lymphopenia. diab 1991;40(11):1375-81
(372)
Yokoi N. Identification of a major gene
responsible for type 1 diabetes in the Komeda diabetes-prone rat. Exp Anim 2005
Apr;54(2):111-5.http://www.ncbi.nlm.nih.gov/pubmed/15897618?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(373)
Yokoi N, Kanazawa M, Kitada K, Tanaka A,
Kanazawa Y, Suda S, et al. A non-MHC locus essential for autoimmune type I
diabetes in the Komeda diabetes-prone rat. J Clin Invest 1997;100:2015-21
(374)
Yokoi N, Hayashi C, Fujiwara Y, Wang HY,
Seino S. Genetic reconstitution of autoimmune type 1 diabetes with two major
susceptibility genes in the rat. Diabetes 2007 Feb;56(2):506-12.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=17259398&query_hl=5&itool=pubmed_docsum
(375)
Komeda K, Noda M, Terao K, Kuzuya N,
Kanazawa M, Kanazawa Y. Establishment of two substrains, diabetes-prone and
non-diabetic, from Long-Evans Tokushima Lean (LETL) rats. Endocr J 1998
Dec;45(6):737-44.PM:10395228
(376)
Yokoi N, Komeda K, Wang HY, Yano H,
Kitada K, Saitoh Y, et al. Cblb is a major susceptibility gene for rat type 1
diabetes mellitus. Nat Genet 2002 Aug;31(4):391-4.PM:12118252
(377)
Kosoy R, Yokoi N, Seino S, Concannon P.
Polymorphic variation in the CBLB gene in human type 1 diabetes. Genes Immun
2004 May;5(3):232-5.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14961073
(378)
Payne F, Smyth DJ, Pask R, Barratt BJ,
Cooper JD, Twells RC, et al. Haplotype tag single nucleotide polymorphism
analysis of the human orthologues of the rat type 1 diabetes genes Ian4
(Lyp/Iddm1) and Cblb. diab 2004 Feb;53(2):505-9.PM:14747305
(379)
Bergholdt R, Taxvig C, Eising S, Nerup
J, Pociot F. CBLB variants in type 1 diabetes and their genetic interaction
with CTLA4. J Leukoc Biol 2005 Apr;77(4):579-85
(380)
Lenzen S, Tiedge M, Elsner M, Lortz S,
Weiss H, Jorns A, et al. The LEW.1AR1/Ztm-iddm rat: a new model of spontaneous
insulin-dependent diabetes mellitus. diabetol 2001
Sep;44(9):1189-96.PM:11596676
(381)
Jorns A, Kubat B, Tiedge M, Wedekind D,
Hedrich HJ, Kloppel G, et al. Pathology of the pancreas and other organs in the
diabetic LEW.1AR1/Ztm- iddm rat, a new model of spontaneous insulin-dependent
diabetes mellitus. Virchows Arch 2004 Feb;444(2):183-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14735361
(382)
Arndt T, Wedekind D, Weiss H, Tiedge M,
Lenzen S, Hedrich HJ, et al. Prevention of spontaneous immune-mediated diabetes
development in the LEW.1AR1-iddm rat by selective CD8+ T cell transfer is
associated with a cytokine shift in the pancreas-draining lymph nodes. diabetol
2009 Jul;52(7):1381-90.PM:19367386
(383)
Jorns A, Gunther A, Hedrich HJ, Wedekind
D, Tiedge M, Lenzen S. Immune cell infiltration, cytokine expression, and
beta-cell apoptosis during the development of type 1 diabetes in the
spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes 2005 Jul;54(7):2041-52
(384)
Lipes MA, Rosenzweig A, Tan KN, Tanigawa
G, Ladd D, Seidman JG, et al. Progression to diabetes in nonobese diabetic
(NOD) mice with transgenic T cell receptors. Science 1993;259(5098):1165-9.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=8267690
(385)
Haskins K, Portas M, Bergman B, Lafferty
K, Bradley B. Pancreatic islet-specific T cell clones from nonobese diabetic
mice. Proc Natl Acad Sci USA 1989;86:8000-4
(386)
Verdaguer J, Yoon J-W, Anderson B,
Averill N, Utsugi T, Park B-J, et al. Acceleration of spontaneous diabetes in
TCR-b-transgenic
nonobese diabetic mice by b-cell cytotoxic CD8+ T cells expressing identical endognous
TCR-a
chains. J Immunol 1996;157:4726-35
(387)
Verdaguer J, Schmidt D, Amrani A,
Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in
monoclonal T cell nonobese diabetic mice. J Exp Med 1997;186:1663-76
(388)
Graser RT, DiLorenzo TP, Wang F,
Christianson GJ, Chapman HD, Roopenian DC, et al. Identification of a CD8 T
cell that can independently mediate autoimmune diabetes development in the
complete absence of CD4 T cell helper functions. J Immunol 2000 Apr
1;164(7):3913-8.PM:10725754
(389)
Wong FS, Visintin I, Wen L, Flavell RA,
Janeway CA. CD8 T cell clones from young nonobese diabetic (NOD) islets can
transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp
Med 1996;183:67-76
(390)
Wegmann DR, Shehadeh N, Lafferty KJ,
Norbury-Glaser N, Gill RG, Daniel D. Establishment of islet-specific T cell
lines and clones from islet isografts placed in spontaneously diabetic NOD
mice. J Autoimmun 1993;6:517-27
(391)
Yang Y, Santamaria P. Dissecting
autoimmune diabetes through genetic manipulation of non-obese diabetic mice.
diabetol 2003 Nov;46(11):1447-64.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14586501
(392)
Haskins K, Wegmann D. Diabetogenic
T-cell clones. diab 1996;45:1299-305
(393)
Chaparro RJ, Burton AR, Serreze DV,
Vignali DA, DiLorenzo TP. Rapid identification of MHC class I-restricted
antigens relevant to autoimmune diabetes using retrogenic T cells. J Immunol
Methods 2008 Jun 1;335(1-2):106-15.PM:18439618
(394)
Holst J, Vignali KM, Burton AR, Vignali
DA. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic
mice. Nat Methods 2006 Mar;3(3):191-7.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=16489336&query_hl=7&itool=pubmed_docsum
(395)
Holst J, Szymczak-Workman AL, Vignali
KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic
mice. Nat Protoc 2006;1(1):406-17
(396)
Arnold PY, Burton AR, Vignali DA.
Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR
retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell
gene transfer. J Immunol 2004 Sep 1;173(5):3103-11.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15322170
(397)
Zhang L, Jasinski JM, Kobayashi M,
Davenport B, Johnson K, Davidson H, et al. Analysis of T cell receptor beta
chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9-23
alpha chains. J Autoimmun 2009 Mar 13.PM:19286348
(398)
Katz JD, Wang B, Haskins K, Benoist C,
Mathis D. Following a diabetogenic T cell from genesis through pathogenesis.
Cell 1993;74:1089-100
(399)
Shi FD, Flodstrom M, Balasa B, Kim SH,
Van Gunst K, Strominger JL, et al. Germ line deletion of the CD1 locus
exacerbates diabetes in the NOD mouse. Proc Natl Acad Sci U S A 2001 Jun
5;98(12):6777-82.PM:11390999
(400)
Masteller EL, Warner MR, Ferlin W,
Judkowski V, Wilson D, Glaichenhaus N, et al. Peptide-MHC class II dimers as
therapeutics to modulate antigen-specific T cell responses in autoimmune
diabetes. J Immunol 2003 Nov 15;171(10):5587-95.http://www.ncbi.nlm.nih.gov/pubmed/14607967?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(401)
Jasinski JM, Yu L, Nakayama M, Li MM,
Lipes MA, Eisenbarth GS, et al. Transgenic insulin (B:9-23) T-cell receptor
mice develop autoimmune diabetes dependent upon RAG genotype, H-2g7
homozygosity, and insulin 2 gene knockout. Diabetes 2006 Jul;55(7):1978-84.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=16804066&query_hl=9&itool=pubmed_docsum
(402)
Kanagawa O, Shimizu J, Vaupel BA. Thymic
and postthymic regulation of diabetogenic CD8 T cell development in TCR
transgenic nonobese diabetic (NOD) mice. J Immunol 2000 May
15;164(10):5466-73.PM:10799914
(403)
Miller JF, Morahan G, Allison J,
Hoffmann M. A transgenic approach to the study of peripheral T-cell tolerance.
Immunol Rev 1991 Aug;122:103-16.PM:1937538
(404)
Oldstone MBA, Nerenberg M, Southern P,
Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes
mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell
1991;65:319-31
(405)
Ohashi PS, Oehen S, Buerki K, Pircher H,
Ohashi CT, Odermatt B, et al. Ablation of "tolerance" and induction
of diabetes by virus infection in viral antigen transgenic mice. Cell
1991;65:305-17
(406)
Apostolou I, von Boehmer H. The TCR-HA,
INS-HA transgenic model of autoimmune diabetes: limitations and expectations. J
Autoimmun 2004 Mar;22(2):111-4.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14987738
(407)
von Herrath MG, Coon B, Lewicki H,
Mazarguil H, Gairin JE, Oldstone MB. In vivo treatment with a MHC class
I-restricted blocking peptide can prevent virus-induced autoimmune diabetes. J
Immunol 1998 Nov 1;161(9):5087-96.PM:9794447
(408)
Laufer TM, von Herrath MG, Grusby MJ,
Oldstone MBA, Glimcher LH. Autoimmune diabetes can be induced in transgenic
major histocompatibility complex class II-deficient mice. J Exp Med
1993;178:589-96
(409)
von Herrath MG, Dockter J, Oldstone MBA.
How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in
a transgenic model. Immunity 1994;1:231-42
(410)
Lo D, Freedman J, Hesse S, Palmiter RD,
Brinster RL, Sherman LA. Peripheral tolerance to an islet cell-specific
hemagglutinin transgene affects both CD4+ and CD8+ T cells. Eur J Immunol 1992
Apr;22(4):1013-22.PM:1348026
(411)
Harlan DM, Hengartner H, Huang ML, Kang
YH, Abe R, Moreadith RW, et al. Mice expressing both B7-1 and viral
glycoprotein on pancreatic beta cells along with glycoprotein-specific
transgenic T cells develop diabetes due to a breakdown of T-lymphocyte
unresponsiveness. Proc Natl Acad Sci U S A 1994 Apr 12;91(8):3137-41.PM:7512724
(412)
King C, Mueller HR, Malo CM,
Murali-Krishna K, Ahmed R, King E, et al. Interleukin-4 acts at the locus of
the antigen-presenting dendritic cell to counter-regulate cytotoxic CD8+ T-cell
responses. Nat Med 2001 Feb;7(2):206-14.PM:11175852
(413)
Apostolou I, Sarukhan A, Klein L, von
Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat
Immunol 2002 Aug;3(8):756-63.PM:12089509
(414)
Morgan DJ, Liblau R, Scott B, Fleck S,
McDevitt HO, Sarvetnick N, et al. CD8(+) T cell-mediated spontaneous diabetes
in neonatal mice. J Immunol 1996;157:978-83
(415)
Vizler C, Bercovici N, Cornet A,
Cambouris C, Liblau RS. Role of autoreactive CD8+ T cells in organ-specific
autoimmune diseases: insight from transgenic mouse models. Immunol Rev 1999
Jun;169:81-92.PM:10450510
(416)
Degermann S, Reilly C, Scott B, Ogata L,
von Boehmer H, Lo D. On the various manifestations of spontaneous autoimmune
diabetes in rodent models. Eur J Immunol 1994 Dec;24(12):3155-60.PM:7528672
(417)
Scott B, Liblau R, Degermann S, Marconi
LA, Ogata L, Caton AJ, et al. A role for non-MHC genetic polymorphism in
susceptibility to spontaneous autoimmunity. Immunity 1994
Apr;1(1):73-83.PM:7889402
(418)
Kurts C, Heath WR, Kosaka H, Miller JF,
Carbone FR. The peripheral deletion of autoreactive CD8+ T cells induced by
cross-presentation of self-antigens involves signaling through CD95 (Fas,
Apo-1). J Exp Med 1998 Jul 20;188(2):415-20.PM:9670055
(419)
Blanas E, Carbone FR, Allison J, Miller
JF, Heath WR. Induction of autoimmune diabetes by oral administration of
autoantigen. Science 1996;274(5293):1707-9
(420)
Heath WR, Allison J, Hoffmann MW,
Schonrich G, Hammerling G, Arnold B, et al. Autoimmune diabetes as a
consequence of locally produced interleukin-2. Nature 1992 Oct
8;359(6395):547-9.PM:1406974
(421) Lesage S, Hartley S, Wilson JD, Goodnow CC. T
cell tolerance in a transgenic mouse model for autoimmune diabetes. Keystone
Symposia Regulation of Immunity and Autoimmunity , 115. 2001.
Ref Type: Abstract
(422)
Wong FS, Dittel BN, Janeway CAJ.
Transgenes and knockout mutations in animal models of type 1 diabetes and
multiple sclerosis. Immunol Rev 1999 Jun;169:93-104
(423)
Elias D, Markovits D, Reshef T, Zee R,
Cohen IR. Induction and therapy of autoimmune diabetes in the non-obese
diabetic (NOD/Lt) mouse by a 65-kDa heat shock protein. Proc Natl Acad Sci USA
1990;87:1576-80
(424) Renold AE, Soeldner JS, Steinke J.
Immunological studies with homologous and heterologous pancreatic insulin in
the cow. Ciba Foundation Colloquia on Endocrinology 15, 122-134. 1964. 1964.
Ref Type: Journal (Full)
(425)
Renold AE, Steinke J, Soeldner JS, Smith
RS, Antoniades HN. Response of calves and heifers to the prolonged
administration of porcine and bovine insulin. XVIIth Scientific Meeting of the
Protein Foundation; 1965 p. 121-2.
(426)
LeCompte PM, Steinke J, Soeldner JS,
Renold AE. Changes in the islets of Langerhans in cows injected with
heterologous and homologous insulin. diab 1966;15(8):586-96
(427)
Renold AE, Steinke J, Soeldner JS,
Antomiades HN, Smith RM. Immunological response to the prolonged administration
of heterologous and homologous insulin in cattle. J Clin Invest
1966;45(5):702-13
(428)
Grodsky GM, Feldman R, Toreson WE, Lee
JC. Diabetes mellitus in rabbits immunized with insulin. diab 1966;15:579-85
(429)
Kloppel G, Freytag G. Insulin antibodies
and immune insulitis in rabbits immunized with bovine or porcine insulin
components. Horm Metab Res 1975;7:25-30
(430)
Glimcher LH, Schroer JA, Chan C, Shevach
EM. Fine specificity of cloned insulin-specific T cell hybridomas: evidence
supporting a role for tertiary conformation. J Immunol 1983 Dec;131(6):2868-74.PM:6196405
(431)
Kahn CR, Rosenthal AS. Immunologic
reactions to insulin: Insulin allergy,
insulin resistance, and the autimmune insulin syndrome. Diab care 1979;2:283-95
(432)
Abiru N, Maniatis AK, Yu L, Miao D,
Moriyama H, Wegmann D, et al. Peptide and MHC specific breaking of humoral
tolerance to native insulin with the B:9-23 peptide in diabetes prone and
normal mice. diab 2001 Jun;50:1274-81.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11375327
(433)
Moriyama H, Wen L, Abiru N, Liu E, Yu L,
Miao D, et al. Induction and acceleration of insulitis/diabetes in mice with a
viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc
Natl Acad Sci U S A 2002 Apr 16;99(8):5539-44
(434)
Liu E, Abiru N, Moriyama H, Miao D,
Eisenbarth GS. Induction of Insulin Autoantibodies and Protection from Diabetes
with Subcutaneous Insulin B:9-23 Peptide Without Adjuvant. Annals New York
Academy of Sciences 2002;958:224-7
(435)
Wen L, Peng J, Li Z, Wong FS. The effect
of innate immunity on autoimmune diabetes and the expression of toll-like
receptors on pancreatic islets. J Immunol 2004 Mar 1;172(5):3173-80.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14978124
(436)
Liu E, Moriyama H, Abiru N, Paronen J,
Devendra D, Finkelman FD, et al. Preventing peptide-induced anaphylaxis:
addition of C-terminal amino acids to produce a neutral isoelectric point. J
Allergy Clin Immunol 2004 Sep;114(3):607-13.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15356565
(437)
Liu E, Moriyama H, Abiru N., Miao D, Yu
L, Taylor RM, et al. Anti-peptide autoantibodies and fatal anaphylaxis in NOD
mice in response to insulin self-peptides B:9-23 and B:13-23. J Clin Invest
2002;110(7):1021-7
(438)
Devendra D, Jasinski J, Melanitou E,
Nakayama M, Li M, Hensley B, et al. Interferon-{alpha} as a Mediator of
Polyinosinic:Polycytidylic Acid-Induced Type 1 Diabetes. diab 2005
Sep;54(9):2549-56.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=16123342
(439)
Graham KL, Sanders N, Tan Y, Allison J,
Kay TW, Coulson BS. Rotavirus infection accelerates type 1 diabetes in mice
with established insulitis. J Virol 2008 Jul;82(13):6139-49
(440)
Paronen J, Liu E, Moriyama H, Devendra
D, Ide A, Taylor R, et al. Genetic differentiation of Poly I : C from B : 9-23
peptide induced experimental autoimmune diabetes. Journal of Autoimmunity
2004;22(4):307-13.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15120754
(441)
von Herrath MG, Guerder S, Lewicki H,
Flavell RA, Oldstone MB. Coexpression of B7-1 and viral ("self")
transgenes in pancreatic beta cells can break peripheral ignorance and lead to
spontaneous autoimmune diabetes. Immunity 1995 Dec;3(6):727-38.PM:8777718
(442)
Pechhold K, Karges W, Blum C, Boehm BO,
Harlan DM. Beta cell-specific CD80 (B7-1) expression disrupts tissue protection
from autoantigen-specific CTL-mediated diabetes. J Autoimmun 2003
Feb;20(1):1-13.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12604308
(443)
Wen L, Chen NY, Tang J, Sherwin R, Wong
FS. The regulatory role of DR4 in a spontaneous diabetes DQ8 transgenic model.
J Clin Invest 2001 Apr;107(7):871-80.PM:11285306
(444)
Allison J, Stephens LA, Kay TW, Kurts C,
Heath WR, Miller JF, et al. The threshold for autoimmune T cell killing is
influenced by B7-1. Eur J Immunol 1998 Mar;28(3):949-60.PM:9541590
(445)
Herrera PL, Harlan DM, Fossati L, Izui
S, Huarte J, Orci L, et al. A CD8+ T-lymphocyte-mediated and CD4+
T-lymphocyte-independent autoimmune diabetes of early onset in transgenic mice.
diabetol 1994 Dec;37(12):1277-9.PM:7534735
(446)
Subudhi SK, Zhou P, Yerian LM, Chin RK,
Lo JC, Anders RA, et al. Local expression of B7-H1 promotes organ-specific
autoimmunity and transplant rejection. J Clin Invest 2004 Mar;113(5):694-700.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14991067
(447)
Serreze DV, Marron MP, DiLorenzo TP.
"Humanized" HLA transgenic NOD mice to identify pancreatic beta cell
autoantigens of potential clinical relevance to type 1 diabetes. Ann N Y Acad
Sci 2007 Apr;1103:103-11. Epub@2007 Mar 21.:103-11
(448)
Macchiarini F, Manz MG, Palucka AK,
Shultz LD. Humanized mice: are we there yet? J Exp Med 2005 Nov
21;202(10):1307-11.PM:16301740
(449)
Fugger L. Human autoimmunity genes in
mice. Curr Opin Immunol 2000 Dec;12(6):698-703.PM:11102775
(450)
Herman AE, Tisch RM, Patel SD, Parry SL,
Olson J, Noble JA, et al. Determination of glutamic acid decarboxylase 65
peptides presented by the type I diabetes-associated HLA-DQ8 class II molecule
identifies an immunogenic peptide motif. J Immunol 1999 Dec 1;163(11):6275-82
(451)
Liu J, Purdy LE, Rabinovitch S, Jevnikar
AM, Elliott JF. Major DQ8-restricted T-cell epitopes for human GAD65 mapped
using human CD4, DQA1*0301, DQB1*0302 transgenic IA(null) NOD mice. diab 1999
Mar;48(3):469-77
(452)
Havari E, Lennon-Dumenil AM, Klein L,
Neely D, Taylor JA, McInerney MF, et al. Expression of the B7.1 costimulatory
molecule on pancreatic beta cells abrogates the requirement for CD4 T cells in
the development of type 1 diabetes. J Immunol 2004 Jul 15;173(2):787-96.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15240665
(453)
Gebe JA, Unrath KA, Falk BA, Ito K, Wen
L, Daniels TL, et al. Age-dependent loss of tolerance to an immunodominant
epitope of glutamic acid decarboxylase in diabetic-prone RIP-B7/DR4 mice. Clin
Immunol 2006 Dec;121(3):294-304.http://www.ncbi.nlm.nih.gov/pubmed/16979383?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum
(454)
Marietta E, Black K, Camilleri M, Krause
P, Rogers RS, III, David C, et al. A new model for dermatitis herpetiformis
that uses HLA-DQ8 transgenic NOD mice. J Clin Invest 2004 Oct;114(8):1090-7.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15489956
(455)
Taneja V, Griffiths M, Behrens M, Luthra
HS, David CS. Auricular chondritis in NOD.DQ8.Abetao (Ag7-/-) transgenic mice
resembles human relapsing polychondritis. J Clin Invest 2003
Dec;112(12):1843-50.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14679179
(456)
Marron MP, Graser RT, Chapman HD,
Serreze DV. Functional evidence for the mediation of diabetogenic T cell
responses by HLA-A2.1 MHC class I molecules through transgenic expression in
NOD mice. Proceedings of the National Academy of Sciences of the United States
of America 2002;99(21):13753-8
(457)
Banuelos SJ, Shultz LD, Greiner DL, Burzenski
LM, Gott B, Lyons BL, et al. Rejection of human islets and human HLA-A2.1
transgenic mouse islets by alloreactive human lymphocytes in immunodeficient
NOD-scid and NOD-Rag1(null)Prf1(null) mice. Clin Immunol 2004
Sep;112(3):273-83.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15308121
(458)
Jarchum I, Takaki T, DiLorenzo TP.
Efficient culture of CD8(+) T cells from the islets of NOD mice and their use
for the study of autoreactive specificities. J Immunol Methods 2008 Nov
30;339(1):66-73.PM:18782577
(459)
Jarchum I, Baker JC, Yamada T, Takaki T,
Marron MP, Serreze DV, et al. In vivo cytotoxicity of insulin-specific CD8+
T-cells in HLA-A*0201 transgenic NOD mice. diab 2007
Oct;56(10):2551-60.PM:17620420
(460)
Shultz LD, Lyons BL, Burzenski LM, Gott
B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in
NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic
stem cells. J Immunol 2005 May 15;174(10):6477-89.http://www.ncbi.nlm.nih.gov/pubmed/17332083?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum1
(461) Brehm MA, Bortell R, Diiorio P, Leif J, Laning
J, Cuthbert A, et al. Human immune system development and rejection of human
islet allografts in spontaneously diabetic NOD-Rag1null IL2rgammanull Ins2Akita
mice. diab 2010 Sep;59(9):2265-70.PM:20570944