Chapter 4
The Role of T-Cells in Beta Cell Damage in NOD Mice and Humans
(Updated 10/07/2011)
Tomasz Sosinowski, Edwin Liu, George Eisenbarth, and Howard W. Davidson
Type 1A diabetes (T1D) is a chronic disorder that results
from the immune-mediated destruction of the insulin-producing ß-cells of the
pancreatic islets1. In its initial phase, which is
clinically silent, T lymphocytes and other inflammatory cells invade the islets
and eventually destroy them. The disease then becomes clinically evident with
the pathological consequences (hyperglycemia, ketosis and long-term
complications) resulting from the inability to maintain glucose and lipid
homeostasis.
Type
1 diabetes is a T-cell mediated disease
The first indication that T1D is an autoimmune disease came from the results of
a comprehensive histological examination of pancreata from diabetic patients
who had died shortly after diagnosis. This showed that most of the subjects had
significant lymphocytic infiltration of their islets concordant with loss of
ß-cell mass 2. Subsequently, islet-cell
antibodies (ICAs) and anti-pancreatic cell-mediated immunity were detected in
recently diagnosed T1D patients 3-5, suggesting that the lymphocytes
accumulated as a result of attraction by antigens derived from pancreatic ß-cells6. Consistent with this hypothesis,
insulitis was only seen in islets containing ß-cells. With the advent of
monoclonal antibodies capable of identifying distinct lymphocyte
sub-populations more detailed immunohistochemical examinations of islet
infiltrates became possible. One of the earliest of such studies showed a
predominance of CD8+ T-cells in the islets of a deceased 12-year old
girl with newly diagnosed T1D, which, together with the observed up-regulation
of MHC class I molecules by islet cells, implicated cytotoxic T-cells (CTLs) in
ß-cell destruction 7. Additional studies of pancreas
from patients with type 1 diabetes have confirmed preponderance of CD8 T-cells
and the presence of B-lymphocytes related to extent of ß-cell destruction 8. The JDRF nPOD (Network for
Pancreatic Organ Donors with Diabetes) program now allows viewing of pancreatic
histology of cadaveric donors directly online (http://www.jdrfnpod.org/).
Similarly, the strong association of T1D with particular MHC II haplotypes (see
chapter 7) suggested a critical role for CD4+ T-cells in the disease
process (reviewed by 9. Additional circumstantial evidence
supporting a crucial role for T-cells and MHC-restricted self antigen
recognition in diabetogenesis came from the reversal and recurrence of diabetes
following twin to twin pancreatic isografts 10,
11, and the inadvertent transfer of
disease between HLA-identical siblings by bone marrow transplantation 12.
The mere presence of T-cells in infiltrates, though highly
suggestive, does not by itself establish a direct role for these cells in the
development of T1D. However, the histological findings were subsequently
followed by reports of T-cell reactivity to ß-cell proteins, providing further
support for the hypothesis. Thus, Roep and colleagues established CD4+ T-cell
lines and clones restricted to HLA-DR from the peripheral blood of new-onset
diabetics after stimulation in vitro
with rat insulinoma cells 13.
Of the eight clones examined, five appeared to recognize insulinoma
membrane components, one of which was a 38kD protein later termed IMOGEN38 14-18. Surprisingly, after expression
cloning IMOGEN38 was shown to be a broadly distributed mitochondrial protein (a
probable subunit of the mitochondrial ribosome), and studies with the human
ortholog suggested that the response was likely xenogeneic (JC Hutton personal
communication). Nonetheless, the identification of several islet cell molecular
targets allowed subsequent studies to be conducted using defined autoantigens,
rather than crude fractions, and have suggested that the peripheral blood of
diabetic subjects and their at-risk relatives contain elevated numbers of
T-cells able to recognize epitopes from ß-cell proteins 19. Such studies suggest that T-cells
provide a legitimate therapeutic target for intervention, and results from clinical
trials of new-onset diabetic patients with
humanized anti-CD3 monoclonal antibodies delayed the deterioration of
circulating C-peptide levels normally seen in the year following diagnosis in 9
of 12 subjects 20-22. Attempts to build upon these
partial successes and improve the therapeutic regimen are currently in
progress.
Animal models of T1D
A. Spontaneous models
Since the target organs of T1D (islets and draining pancreatic lymph nodes) are
inaccessible in human subjects, the study of T1D has been greatly facilitated
by the availability of animal models such as the Biobreeding-diabetes prone
(BB-DP) rat 23 and the nonobese diabetic (NOD)
mouse 24, which spontaneously develop
diseases that mimic many features of human T1D. In particular, the NOD mouse has
been the subject of extensive studies for over 20 years 25-27, and has provided key experimental
evidence supporting several strategies to treat the human disease, some of
which are showing promise in initial clinical trials 28,
29. Numerous abnormalities have been
reported in the immune systems of NOD mice, including defects in antigen
presenting cells (APCs) 30 and hyporesponsiveness of T
lymphocytes 31,
32, which together may compromise both
central 33 and peripheral 34 tolerance to pancreatic ß-cells.
Similar abnormalities have been reported in human subjects (for example 35-37, and it has been proposed that
T-cell hyporesponsiveness may be a general feature conferring susceptibility to
inflammatory autoimmune disorders 38. However, it must be noted that the
immune systems of humans and mice show several key differences (reviewed by 39, which must be kept in mind when
extrapolating between these species 28,
40-42. Indeed, there are notable
differences between T1D in NOD mice and humans, not the least being that NOD
mice only develop disease if kept in specific pathogen free conditions.
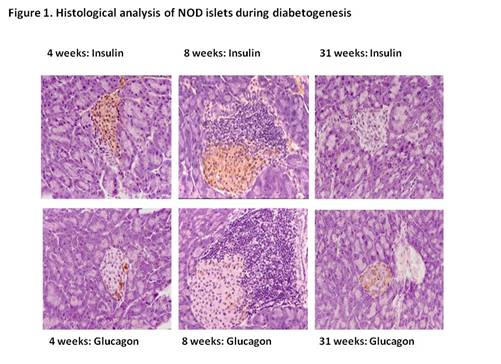
Moreover, in these animals disease is associated with pronounced cellular
infiltrates that surround the individual islets (Figure 1) that begin to form
at least 7 weeks prior the onset of overt
disease 43,
44. In contrast, pancreata obtained at
post mortem from diabetic subjects who died shortly after clinical
manifestation of T1D typically show a much less florid infiltration (e.g. 7,
45). In that NOD mice are inbred they
can only be considered to be the equivalent of a single genotype "case
study" of T1D and a genotype that is homozygous at all loci.
Although
the precise sequence of events that lead to T1D in NOD mice remain uncertain,
the central role of T-cells in diabetogenesis in these animals is
incontrovertible. Thus, treatment of newly diabetic animals with anti-CD3
antibodies, which suppress immune responses by transient T-cell depletion and
modulation of T-cell Receptor (TCR) signaling, induces long-term remission 46,
47.
Moreover, diabetes can be transferred to immuno-compromised hosts by
mixed T-cell populations, or in some instances, individual T-cell clones 48-51. For example, Haskins
and colleagues isolated 8 CD4+ T-cell clones recognizing islet
antigens (including chromagranin and islet amyloid polypeptide) 52,
53 presented by the NOD MHC class II
molecule, I-Ag7, at least 5 of which are capable of inducing disease
after adoptive transfer 54,
55. Transgenic mice expressing the
T-cell receptor (TCR) of one of the diabetogenic clones, BDC2.5 (target peptide
derived from chromogranin), have been generated and bred onto the NOD
background 56. Interestingly, evaluation of the
lymphoid compartment in NOD/BDC2.5 animals showed no sign of negative selection
in the thymus, with normal peripheral T-cell reactivity. This was also true of
transgenic C57BL/6-H-2g7 (B6g7/BDC2.5) animals. However,
autoimmune pathogenesis was highly dependent upon the genetic background of the
animal 57. In both NOD and B6g7
transgenics there was no manifestation of disease in the first 2 weeks of life,
with pancreatic islets completely free of infiltration. Insulitis appeared
abruptly at 18 days, and subsequently progressed to eventually involve
essentially all islets. However, whilst the B6g7/BDC2.5 animals
suffered a highly aggressive insulitis and the majority rapidly progressed to
overt disease, paradoxically, NOD/BDC2.5 animals were significantly protected
from spontaneous disease, and exhibited a more benign "respectful"
insulitis. Nevertheless, young
NOD/BDC2.5 are significantly more sensitive to cyclophosphamide induced
diabetes than their non-transgenic relatives 58, and NOD/scid/BDC2.5 mice rapidly progress to overt T1D and typically die of
diabetic complications on or before 33 days of age 59. The precise mechanisms by which
the un-manipulated NOD/BDC2.5 animals restrain their insulitis are currently
uncertain, but may involve populations of T-cells expressing alternative a T cell
receptor chains due to incomplete allelic exclusion at this locus 56,
60.
In
addition to TCR transgenic animals, targeted gene disruption and retrogenic 61 technologies have also been used to
study other features of T1D in NOD mice. For example, mice lacking expression
of ß2-microglobulin (ß2M), that consequently do not
express functional MHC class I molecules, do not develop insulitis 62-65, implicating CD8+
T-cells in the initiation of disease. Interestingly, transgenic restoration of
MHC class I expression in NOD-ß2M-/- mice using different
ß2M alleles provided contrasting results. Thus, animals
reconstituted with the endogenous ß2Ma allele developed
T1D, whilst those given the ß2Mb allele (which only
differs at a single amino acid residue) did not 66. At present the precise mechanism
of protection is uncertain, although it may reflect subtle differences in the
peptide-binding properties of the resultant MHC class I molecules 67.
Targeted
gene disruption has also provided evidence for a key role for proinsulin in
diabetogenesis in NOD mice. In contrast to humans, mice have 2 non-allelic
insulin genes (insulin 1 on chromosome 19, and insulin 2 on chromosome 7), and
recently NOD mice with disruptions in either one have been created 68,
69. Surprisingly, the mice show
contrasting phenotypes; insulin 1-/- mice are markedly
protected from diabetes (but do develop anti-insulin autoantibodies), while
insulin 2-/- animals show accelerated disease. In contrast, animals
whose sole preproinsulin is a mutant form of insulin 2 in which the
immunodominant B:9-23 epitope is disrupted are completely protected from
diabetes 70, although they do develop sialitis
confirming the organ-specificity of the effect. Such results are at variance
with those from related studies in which NOD mice lacking the autoantigens
GAD65, IA-2, phogrin (IA-2ß) and IGRP 71 were produced and where disease
occurrence was not significantly altered 72-74. This suggests a central role for
(pro) insulin in the disease process, although the precise mechanisms of
acceleration, or protection from disease, remain to be determined. The
potential central role of insulin and insulin peptide B:9-23 is supported by
studies where both insulin genes are eliminated and an insulin mutated at
position B16 (Y to A). These mice do not develop diabetes 70,
75. Krishnamurthy and coworkers have
found that eliminating response to proinsulin eliminates the prominent CD8
T-cell response to IGRP 76,
77. Vignali and coworkers have
introduced a new methodology to study T-cell receptor targeting of islet ß
cells, namely the creation of retrogenic mice 78,
79. The creation of retrogenic mice
utilizes retroviruses to introduce T-cell receptors into bone marrow cells that
are then transplanted into immunodeficient mice. This technology greatly
accelerates studies of such T-cell receptors compared to creating transgenic
mice. Within 8 weeks, the pathogenicity of T-cell receptors can be assessed.
Studying a series of 17 retrogenes, those targeting GAD failed to induce
diabetes. Insulin peptide B:9-23 reactive TCR caused delayed diabetes and
IA-2/phogrin TCR caused insulitis and TCRs targeting chromogranin (e.g. BDC
2.5) caused diabetes as did TCR’s targeting unknown molecules. (Diabetes
induction TCR: BDC-10.1 > BDC-2.5 > NY 4.1 > BDC 6.9). Of note, a series of TCRs targeting GAD65
caused fatal encephalitis independent of the induction of anti-GAD
autoantibodies61.
No insulitis was observed.
Presumably this was related to the lack of GAD in mouse islets.
B. Experimental autoimmune diabetes
(EAD)
As
only a limited number of animal models exhibiting spontaneous disease are
currently available, systems to induce EAD in non-autoimmune prone mice have
also been developed. Typically these are based upon the transgenic ß-cell
expression of heterologous proteins under control of the rat insulin promotor
(RIP). Such models have provided key insights into the establishment and breaking
of tolerance to ß-cell antigens. Initial studies showed that some lines of
RIP-Tag C57BL/6 mice, which express the SV40 large T antigen (Tag) in their
ß-cells, were intolerant of the transgene, developing spontaneous autoimmunity 80. Tolerance, or autoimmunity,
correlated with the presence or absence of embryonic expression of the
transgene, with animals that did not express Tag until adulthood developing
disease. Moreover, tolerant animals also expressed the transgene in the thymus 81, suggesting that, at least for some
proteins, central tolerance can be established to apparently organ-specific
antigens (reviewed in 82. However, central tolerance could
be broken if the precursor frequency of peripheral autoreactive T-cells was too
high. Thus, tolerant RIP-Tag mice crossed with transgenic mice showing low
expression (~10% of peripheral T-cells) of the TCR of a Tag specific CTL were
protected from spontaneous disease 83. In contrast, offspring of parents
showing high expression (~90%) of the transgenic TCR were intolerant. The importance of central tolerance was also
demonstrated in a model where CBA (H-2k) mice expressing the Kb
MHC class I molecule under control of the RIP were crossed with transgenic mice
expressing the TCR of an H-2k-restricted CTL clone recognizing Kb.
In this case the progeny rejected skin grafts from H-2b animals, but
were protected from T1D. However, crossing the double transgenic mice with
RIP-IL-2 animals caused rapid onset disease 84. Neonatal replacement of the
thymuses of the double transgenic animals with tissue from non-transgenic mice
permitted high avidity Kb-specific T-cells to reach the periphery,
which allowed disease to be triggered in animals primed either with allogeneic
skin grafts or the injection of irradiated splenocytes from H-2b
donors 85. Some animals required multiple
primings, suggesting that the duration of the stimulus, as well as the avidity
of the peripheral autoreactive T-cells, profoundly influences the course of
disease.
The
lack of spontaneous disease in RIP-Kb x Kb-TCR mice after
thymus replacement, despite the presence of peripheral high affinity Kb-specific
T-cells, indicates that protective mechanisms exist in the periphery which can
act to negate a lack of central tolerance. Two potential mechanisms have been
proposed, namely immune ignorance and active tolerance 86, and evidence that both are
involved in protection from T1D has come from RIP-based transgenic models. For
example, two strains of mice secreting either high (RIP-Ovahi) or
low (RIP-Ovalo) levels of ovalbumin from their ß-cells were exposed
to 5, 6-carboxy-succinimidyl-fluorescein-ester (CSFE) labeled OT-1 T-cells
specific for ovalbumin. Analysis of the pancreatic draining lymph nodes 3 days
after transfer revealed that the OT-1 cells had proliferated in the RIP-Ovahi
but not the RIP-Ovalo mice 87, indicating that the level of
antigen acquired by antigen presenting cells (APCs) in the RIP-Ovalo
animals was insufficient to trigger naive T-cells, and consequently that the immune
system was ignorant of the presence of the neo-antigen. Both strains developed
T1D if exposed to pre-activated OT-1 CTLs, confirming that the RIP-Ovalo
animals did indeed exhibit ß-cell expression of the transgene. However, despite
proliferation in the draining lymph nodes, adoptive transfer of even 107
naive OT-1 cells to RIP-Ovahi mice did not induce disease 88, suggesting that effector CTL
generation was inefficient under these conditions. Nonetheless, tolerance to
ß-cell expression of ovalbumin could be broken if the efficiency of
presentation was enhanced. Thus, studies with RIP-mOva mice, which express a
membrane-anchored chimeric fusion protein derived from ovalbumin and the
transferrin receptor 89, demonstrated that cell-associated
ovalbumin was cross-presented to CD8+ T-cells ~50,000-fold more
efficiently than the secreted form 90and adoptive transfer of 5 x 106
naive OT-1 T-cells into non-irradiated
RIP-mOva mice rapidly induced disease.
To
analyze the fate of OT-1 cells following transfer into RIP-mOva mice without
inducing disease these animals were crossed with bm1 mice (that have a mutation
in the Kb molecule which prevents presentation to OT-1 T-cells 91. The resulting RIP-mOva.bm1 mice
were lethally irradiated and reconstituted with bone marrow from wild-type
C57BL/6 animals so that OT-1 cells could interact with hematopoietic, but not
peripheral, cells. Analysis of draining lymph nodes 3 days after adoptive
transfer of CSFE-labeled OT-1 T-cells confirmed that they had proliferated, but
after 8 weeks transgenic T-cells comprised only ~1% of CD8+ T-cells
recovered from the spleens and lymph nodes of B6®RIP-mOva.bm1 mice as compared to
~7.4% in B6®bm1 animals, suggesting that activation had led to
peripheral deletion92. Further studies indicated that
deletion was dependent upon CD95 (Fas) expression by the OT-1 cells, and that
surface expression of CD95 was up-regulated following cross-presentation in the
draining lymph nodes 93. However, the presence of
antigen-specific CD4+ T-cells impaired deletion of activated CTLs,
and instead led to enhanced activation and expansion. Thus, neither 2 x 106
OT-II cells (a CD4+ ovalbumin-specific clone), nor 2.5 x 105
OT-I cells, by themselves were diabetogenic in RIP-mOva mice. In contrast, 68%
of animals receiving both populations of cells became diabetic within 10 days
of transfer 94, which corresponded with an
~10-fold increase in the numbers of OT-1 cells in their spleens and lymph nodes
after 4 days as compared to animals that did not receive OT-II cells.
Interestingly, experiments using mixed chimeras of bone marrow cells from I-A-/-,
I-A-/-CD40-/-, and bm1 mice demonstrated that it was not
necessary for the same APC to activate both the OT-I and OT-II cells, but that
T1D only resulted when the APC interacting with the naive OT-I cells had been
activated via CD40 by activated OT-II cells expressing CD154 (CD40 ligand).
Antigen-antibody immune complexes can also "license" dendritic cells
to efficiently prime CTLs 95, suggesting a potential mechanism
by which autoantibodies might contribute to the expansion of the autoimmune
response.
The
RIP-Ova based studies indicate that efficient mechanisms exist to maintain
tolerance to ß-cell proteins, but that these can be overcome if the peripheral
frequency of high avidity T-cells exceeds a critical threshold, by co-operation
between CD4+ and CD8+ T-cells, or by local expression of
pro-inflammatory cytokines. Similar conclusions were reached in another EAD
model based upon expression of the glycoprotein (GP) or nucleoprotein (NP) of
lymphocytic choriomeningitis virus (LCMV). Thus, RIP-LCMV mice do not develop
spontaneous diabetes, but disease is triggered by LCMV infection 96,
97. In contrast, spontaneous disease
occurs in double transgenic mice co-expressing RIP-NP and RIP-IFNa 98, or RIP-GP and RIP-B7.1 99. As with the RIP-Tag mice, thymic
expression of the LCMV proteins was not observed in all founders, which
significantly influenced the rate and characteristics of disease in their
progeny. Animals lacking thymic expression of the transgene rapidly developed
T1D in a CD4+-independent manner following infection, whilst thymic
expression significantly slowed disease induction, which in this case was
dependent upon the presence of both CD8+ and CD4+ T-cells
100. RIP-NP x RIP-B7.1 double
transgenic mice that had thymic expression of the viral protein and also
express CD80 (B7.1) on their pancreatic ß-cells, do not develop spontaneous
disease, although T1D is accelerated in these animals following viral infection
relative to their RIP-NP single transgenic littermates. Thymic expression of
the transgene caused negative selection of high avidity CD8+ T-cells
specific for the immunodominant epitope of NP 101, although interestingly, the viral
infection was cleared with essentially identical kinetics in the presence or
absence of negative selection. Comparison of the T-cell responses to LCMV in
RIP-NP and non-transgenic animals revealed a significant skewing of the
anti-viral response towards a normally sub-dominant epitope in NP, suggesting
that high avidity T-cells to sub-dominant or cryptic epitopes within
autoantigens can escape negative selection. This may have important
implications during the expansion of the auto-response in T1D.
In
both CD4+-dependent and independent RIP-LCMV models T1D induction
depends upon infection with a virus capable of causing pancreatic inflammation 102, highlighting a key role of viral
trophism, and consequent local exposure to pro-inflammatory chemokines and
cytokines, in overcoming peripheral tolerance to islet antigens in this model.
Consistent with this conclusion immunization of RIP-LCMV mice with an LCMV-GP
peptide caused expansion of autoreactive CTLs but did not induce T1D unless TLR
ligands such as the viral mimic polyinosinic-polycytidylic acid (poly I/C) were
co-administered 103. Similarly, co-expression of
IL-2 104 or the chemokine CXCL10 (IP10) 105in islets does not induce
spontaneous disease, but enhances T1D development after initiation of the
anti-self response. However, genetic factors influence the degree of
"pre-lesioning" necessary to allow activated autoreactive CTLs to
infiltrate islets and destroy ß-cells 106, suggesting that in at least some
individuals susceptibility to T1D in humans likely includes
hyper-responsiveness to pro-inflammatory stimuli.
Experiments
using a panel of closely related viruses demonstrated a critical threshold for
the frequency of high avidity cross-reactive CTLs that must be exceeded to
induce disease, and showed that variations between the primary sequences of the
ß-cell protein and its viral mimic either in the flanking residues of the
immunodominant epitopes that effect their processing in APCs, or in the
epitopes themselves, significantly influences whether or not this occurs.
Interestingly, experiments conducted with combinations of LCMV and the
distantly related Pichinde virus (PV), which mimics a sub-dominant epitope in
LCMV, suggested that a mimicking low-affinity epitope could re-activate resting
antigen-experienced CTLs under conditions where it did not activate naive CD8+
T-cells 107. Thus, exposure to PV alone did not
induce disease in thymically-expressing RIP-NP mice, nor did it accelerate T1D
in those animals subsequently given LCMV. In contrast, exposure to PV following
prior infection with LCMV significantly enhanced the rate of onset of diabetes.
This suggests that the order of exposure to two potentially auto-reactive
viruses can significantly influence the nature of the immune response to self,
and that disease might be triggered by reactivation of memory cells generated
during a previous sub-threshold response to a potentially diabetogenic virus by
a subsequent infection with a cross-reactive but otherwise non-diabetogenic one
108.
In
contrast to the rapid-onset RIP-LCMV model, exposure of adult Ins-HA mice
(whose ß-cells, but not thymuses, express the influenza virus strain A/PR/8/34
hemagglutinin (HA) at high levels) to cognate virus did not induce disease 109. Although this may in part be due
to the fact that this strain of influenza is unable to replicate in mice, there
is also evidence that it represents the establishment of dominant peripheral
tolerance to the neo-antigen following efficient cross-presentation of the
antigen prior to immunization 110,
111, possibly via the expansion of
antigen-specific regulatory or suppressor cells. Immunization of neonatal
Ins-HA animals with influenza virus rapidly caused T1D 112, consistent with the observation
that constitutive, but not inflammatory, cross-presentation is disabled in the
pancreas of young mice 113. Similarly, rapid onset spontaneous
disease was observed in H-2d Ins-HA mice crossed with mice
transgenic for the TCR of the Kd-restricted HA-specific CTL clone 4 114. Spontaneous T1D also occurred in
approximately 30-40% of Balb/c (H-2d) Ins-HA mice crossed with
animals transgenic for the TCR of the I-Ed-restricted HA-specific
clone 6.5 115, but not in the same mice crossed
with HNT-TCR transgenic animals that recognize HA126-138 in the
context of I-Ad 116. However, HNT-TCR T-cells are diabetogenic
in B10.D2 Ins-HA mice, highlighting the importance of non-MHC genes in
determining whether a pathogenic or protective response occurs. Peripheral
tolerance is not always effective, as evidenced by the spontaneously diabetic
RIP-Tag mice, and by the fact that, in contrast
to the InsHA mice (none of whom develop
spontaneous disease), a significant proportion (13-27%) of mice expressing the
HA of influenza A/Japan/305/57 under control of the RIP developed spontaneous
T1D 117.
EAD
has also been induced using natural diabetic autoantigens. Thus, immunization
with a peptide from the insulin B chain caused the development of insulin
autoantibodies, but not insulitis in Balb/c (H-2d), but not C57/BL6
(H-2b), mice 118. Co-administration of peptide and
poly I/C to Balb/c mice led to a predominantly CD4+ insulitis, but
not hyperglycemia 119. However, immunization of RIP-B7.1
transgenic mice on an H-2d/b or H-2d/d background with
either poly I/C or the insulin peptide alone induced autoimmune diabetes, which
was accelerated by co-administration of both agents. The mechanism(s)
underlying disease induction are incompletely understood, and may vary slightly
for the peptide and mimic. Given that under these circumstances the insulitis
was predominantly comprised of CD8+ T-cells, and expression of B7.1
on pancreatic ß-cells abrogates the requirement for CD4+ T-cells in
diabetogenesis 120, it appears likely that the peptide
acts to promote expansion/activation of insulin-specific CD8+
T-cells similar to the H2-Kd-restricted clone G9C8 121. In contrast, since poly I/C will
induce the production of type 1 interferons by APCs, disease induction by the
viral mimic is probably mechanistically similar to that induced by transgenic
islet expression of IFNa 122, with bystander activation of
APCs 123 causing expansion of pre-existing
auto-reactive CTLs 124,
125. T1D induction by poly I/C probably
also involves direct stimulation of islet cells via TLR3 and consequent
secretion of chemokines such as IP-10 126, and will likely produce a more
diverse initial CD8+ response than produced with the peptide,
possibly including ß-cell proteins such as IGRP as well as insulin. In each
case ß-cell expression of the co-stimulatory molecule will promote
amplification of the response within the target organelle, and consequent
destructive insulitis. Bystander activation has also been proposed as the
underlying mechanism in disease induction by coxsackie virus B4 in NOD/BDC2.5
transgenic mice 127, and is likely to be important in
the accelerated T1D observed in NOD/RIP-B7.1 animals 128, and the diabetes observed in
RIP-B7.1 x RIP-TNFa double transgenic mice 129. EAD can also be induced in H-2b/b
RIP-B7.1 mice by vaccination with (pro) insulin cDNA, but not GAD65 cDNA 130, further highlighting the
importance of (pro) insulin in the initiation of disease.
Checkpoints in the development of autoimmune diabetes
Studies with NOD/BDC2.5 TCR transgenic mice and various congenic NOD strains
have allowed investigators to define three major checkpoints in the
pathogenesis of T1D 131. Checkpoint 0 controls the
development of an autoimmune prone T-cell repertoire, checkpoint 1 the onset of
insulitis, and checkpoint 2 the switch from controlled insulitis to overt
diabetes (Figure 2). Each appears to be regulated by multiple susceptibility
loci132 and ill-defined epigenetic or
environmental factors, such that passage through checkpoints 0 and 1 does not
inevitably lead to clinical disease, and even an extensive and active insulitis
can persist for long periods of time without significant ß-cell depletion. For example, congenic
NOD.B10 Idd9 mice develop an aggressive insulitis and high insulin autoantibody
expression133 but rarely progress to diabetes.
Although the natural history of human T1D is temporally much more variable than
the mouse models, with symptomatic disease occurring at any time from the first
year of life to well into middle-age, and does not show the same pronounced sex
bias observed in most NOD colonies, it is believed that it also involves
passage through similar checkpoints that mark key changes in the autoimmune
process.
The
molecular and cellular mechanisms that underlie these checkpoints are not just
of academic interest since therapeutic interventions to prevent, or arrest
human T1D prior to complete ß-cell destruction, may stem from influencing such
control mechanisms. Moreover, the multiplicity of factors that regulate disease
progression suggest that distinct therapeutic strategies are likely to be
required to treat individuals at the various stages of T1D. Consequently they
are currently the focus of considerable research. It has long been appreciated
that the specific MHC class II alleles expressed by an individual are a crucial
factor in controlling whether or not checkpoint 0 is passed (see Chapter 7),
and that expression of alternative alleles in autoimmune prone mice can have a
dominant protective effect. At present
the precise mechanisms by which particular alleles increase or decrease
susceptibility to specific autoimmune diseases (e.g. DRB1*1501-DQB1*0602
haplotypes confers susceptibility to multiple sclerosis but protection from
T1D) remains unresolved, although this presumably reflects the nature of the
trimolecular complex interaction between the MHC class II bound autoantigenic
peptide and TCR of autoreactive T-cell. In this regard it is interesting to
note that the recent crystal structures of 3 autoimmune TCRs bound to their
cognate ligands showed unconventional interactions resulting in sub-optimal
contact with the MHC bound self-peptide134-137. This would presumably lead to a
low functional avidity and increased likelihood of a failure of negative
selection due to a consequent resistance to activation-induced apoptosis 138,
139, especially if molecular defects in
pro-apoptopic pathways were also present 140. However, it is now recognized that
autoreactive T-cells can also be protective and that the same peptide/MHC class
II glycoprotein combination can stimulate both diabetogenic and non-pathogenic
T-cells141,
142, consistent with the notion that
the intrinsic balance between protective and pathogenic T-cell subsets in an
individual is only partly dependent upon their MHC haplotype.
Figure 2. Probable checkpoints in
the development of T1D
Checkpoint 0
Genetic
pre-disposition (hyporesponsive T-cells, hyper- or hypo-responsive APCs,
ß-cells hypersensitive to stress induced apoptosis)
Reduced
negative selection of high avidity ß-cell specific T-cells and/or insufficient
generation of ß-cell specific regulatory T-cells
Inherent
Th1 v Th2 bias
![]()
No Insulitis
Risk-conferring peripheral T-cell repertoire
Checkpoint
1
Proinflammatory
environment in pancreas and draining pancreatic lymph nodes (Infection or
ß-cell necrosis)
Activation
of ß-cell specific CTLs and Th1 biased CD4+ T-cells
![]()
Controlled
Insulitis
Sufficient intra-islet amounts of
protective cytokines and/or regulatory T-cells to prevent uncontrolled ß-cell
apoptosis
ß-cells retain functional and
recovery/replicative capacity
Checkpoint 2
Avidity
maturation of autoresponse and expansion via epitope spreading
Recruitment
of additional effector cells
Breakdown
of protective mechanisms (decrease in intra-islet anti-inflammatory
chemokines/cytokines and regulatory cells)
Cytokine,
oxidative, and/or hyperglycemic stress on ß-cells and loss of functional and
recovery/replicative capacity
Uncontrolled
ß-cell apoptosis (cell contact and/or stress dependent)
![]()
Destructive Insulitis and functional
ß-cell insufficiency
Overt T1D
Figure 4.2. Probable checkpoints in the development of TID
The
virtually co-incident temporal onset of peri-insulitis in NOD/BDC 2.5 TCR transgenic
animals (where checkpoint 0 is by-passed), and their non-transgenic relatives,
indicates that the observed 2-3 week delay does not represent the time required
to establish an expanded auto-reactive peripheral repertoire per se, but rather reflects an inherent
difference in the homing potential of the T-cells and/or “attractiveness” of
the islets. This is likely to be due to multiple factors including increased
expression of lymphocyte addressins (especially MadCAM-1 and PNAd) by the
pancreatic blood vessel endothelium 143-145, chemokine-dependent activation of
their ligands (ega4ß7 integrin 146-148) on the surface of activated
lymphocytes (reviewed by149, and presentation of islet antigens
by endothelial cells 150,
151. Together such changes will promote
extra-vascularization of activated auto-reactive T-cells within the islet,
although it remains uncertain precisely how such events are triggered. In this
regard it should be noted that three immunologically important events coincide
with the passage of NOD mice through checkpoint 1; weaning, a wave of islet
cell apoptosis due to tissue remodeling, and the establishment of a
preferential trafficking route from the gut to the pancreatic draining lymph
nodes 152. The former is associated with
significant changes in the intestinal flora and exposure to a large array of
novel food antigens. This transiently induces a broad T-cell stimulation,
likely involving both mesenteric and pancreatic lymph nodes, which in
susceptible animals could lead to bystander damage of islets, or direct targeting
of pancreatic ß-cells due to molecular mimicry 131,
152.
Weaning
is also associated with increased ß-cell activity due to the switch to a more
carbohydrate-rich diet. This probably renders the islets more susceptible to
stress-related damage 153, which in pre-disposed individuals
could trigger the expression of the pro-apoptopic death receptor 4 ligand by
stressed ß-cells and their subsequent killing and phagocytosis by macrophages 154, thereby providing a source of
antigens for presentation to auto-reactive T-cells 155.
Similarly, the wave of remodeling-induced islet cell apoptosis could
also provide a source of ß-cell antigens for presentation by APCs 156,
157. All of these events occur in both
resistant, and diabetes prone animals, suggesting that they are not inherently
diabetogenic. Indeed in normal circumstances they will probably result in
peripheral tolerance 157,
158, but in the context of intestinal
stress or injury (for example due to a viral infection 159 and/or intrinsic abnormalities in
APCs and lymphocytes such as are observed in NOD mice 160,
161, may lead to an immunogenic rather
than tolerogenic response. The mouse models also indicate that the timing of
exposure to a potential autoantigen during the development of the immune system
during neonatal life can be critical in determining whether a protective or
immunogenic response results, and interestingly, recent research has suggested
that there may be a time window in infancy outside which initial exposure to
cereals increases T1D risk in susceptible children 162.
The autoantigens
At
present the specificity, and antigenic diversity, of the diabetogenic T-cells
whose activation at checkpoint 1 initiates the process that ultimately leads to
disease remains uncertain. Given the tissue specificity of T1D it is tempting
to speculate that the primary targets are ß-cell specific and proinsulin is a
prime target with ability of islets containing the B:9-23 epitope when
transplanted to activate disease. Multiple other targets are present including
peri-islet Schwann cells 163. Similarly, although activation of
T-cells in draining pancreatic lymph nodes is clearly critical for
pathogenesis, and surgical removal of these structures from young (3 week old)
NOD mice prevented T1D in at least 80% of the treated animals 164, a recent study unexpectedly showed
that lymphocytes isolated from
mesenteric lymph nodes of 3 week-old NOD mice were almost 4-fold more
diabetogenic than those from the pancreatic nodes of the same animals, following
adoptive transfer into NOD.scid recipients 165. Consistent with previous studies,
Jaakkola and colleagues also showed that, in contrast to the 3-week old
animals, at 6 weeks of age lymphocytes from the pancreatic nodes were the most
effective in transferring disease, whilst at later times the spleen contained
the highest proportion of diabetogenic cells. Such results might indicate the
presence of a significant proportion of regulatory cells in the pancreatic
nodes of the 3 week-old animals 166, but instead could also suggest
that the initial priming is to a foreign antigen that occurs in the
gut-associated tissue, and that this response is subsequently amplified and
expanded to include islet cell antigens in the pancreatic nodes. This latter
hypothesis is consistent with the previously observed effects of blocking the
mucosal addressin MAdCAM-1 167, and is not inconsistent with the
fact that at early time points activated BDC2.5 TCR transgenic T-cells are only
found in islets and their draining lymph nodes 168, if it is assumed that in a
non-transgenic NOD mouse the equivalent cells are activated as part of the
presumptive amplification stage, given that their cognate antigen is believed
to be islet-specific 169. Thus it is possible that in NOD
mice passage through checkpoint 1 occurs due to the temporal co-incidence of a
wave of islet cell apoptosis that leads to enhanced presentation of ß-cell
antigens in the pancreatic lymph nodes, and exposure to novel antigens in the
gut that activate APCs and/or Th1 polarized T-cells that subsequently migrate
to the pancreas and perturb the response in the draining nodes such that an
immunogenic response to islet cell antigens results. In the transgenic animals
the wave of apoptosis alone would be sufficient to activate the diabetogenic
cells due to their high frequency and the lack of appropriate regulatory cells,
and so molecular mimicry would not be required. A similar involvement of
post-natal islet cell apoptosis 170, congenital ß-cell abnormalities, and dietary 162,
171
or enteroviral triggers, in human T1D have also been proposed, although
at present their relative roles (if any), and generality as casual factors,
remain controversial.
Nonetheless, whether the auto-immune response is initiated
directly, or as a secondary consequence of a primary reaction to a foreign
antigen, it is clear that islet cell antigens are critical to the disease
process 172, and that a detailed knowledge of
their molecular characteristics is essential both to the rational design of
immunotherapies, and in the monitoring of at-risk individuals. Moreover,
although disease can be driven by T-cells having single specificities in
immuno-compromised animals, and there may be a restricted number of islet-cell
reactive clones in early pancreatic infiltrates in NOD mice 173,
174, natural disease progression
appears to involve multiple autoantigens 175 and both epitope spreading within 176, and avidity maturation of 177, the T-cell response. As T-cells
with identical specificities can adopt either pathogenic or tolerogenic
properties142,
178, and a monoclonal regulatory T-cell
population can suppress a polyclonal diabetogenic response179-181, it is likely that any diabetic
autoantigen has the potential to be used therapeutically. Thus, considerable
effort has been devoted to identifying the molecules themselves, and the
epitopes within them, that interact with particular MHC glycoproteins. To date
the majority of diabetic autoantigens that have been defined at the molecular
level were discovered either by a candidate gene approach, or by the
identification of antibody targets in human diabetic sera. Insulin, the 65
kilodalton form of glutamic acid decarboxylase (GAD65), the insulin granule
membrane proteins ICA512 (IA-2), phogrin (IA-2ß) and ZnT8 are major targets of
circulating islet autoantibodies in man.
Of these only insulin appears to be ß-cell specific and ZnT8 islet
specific, whereas the others are broadly distributed among neuroendocrine
tissues such as the brain, pituitary and adrenal medulla. The humoral response per se probably contributes little to
the pathogenesis of the disease of man 182, and although B-lymphocytes may be
important for antigen presentation in T1D 183, they are not essential 184. Nonetheless, circulating
autoantibodies provide useful pre-clinical markers for diabetic autoimmunity,
and may also play a role in modulating T-cell responses through their effects as
APCs. ZnT8, the islet zinc transporter, is the most recently identified target
of humeral immunity of man and studies of T-cells reacting with ZnT8 are
underway 185,
186.
Given that the production of high affinity antibodies is a
T-dependent process, it is reasonable to suggest that molecules recognized by
autoantibodies might also be the targets of autoreactive T-cells, although this
need not be the case for particulate antigens due to the process of linked
activation. Nonetheless, this hypothesis appears correct, at least for (pro)insulin,
IA-2/phogrin and GAD65 187 and specific CD4+ and
CD8+ T-cell clones have been isolated from spleen, lymph nodes or
islet infiltrates of pre- or newly diabetic NOD mice 54,
188-190 which do not correspond to any of
the known serological markers. Similarly, HLA-DR restricted CD4+
T-cell lines have been isolated from new onset T1D patients that recognize
currently unidentified ß-cell antigens 191. This may indicate that their
target antigen is a presently unidentified component of ICAs, or is
intrinsically unable to elicit a humoral response, or in the case of CD8+
cells, the targeting of epitopes not present in a stable protein 192,
193.
Defining the cognate antigens for these “orphan” clones remains an
important goal, and with the recent advances in proteomic techniques, now
appears a realistic aim. Thus, using a combination of a sensitive bioassay and
high-end liquid chromatography coupled to tandem mass spectrometry to analyze
peptides eluted from H-2Kd molecules from NIT1 insulinoma cells the
target of the well studied NY8.3 CD8+ clone was shown to be a peptide derived
from the ß-cell protein islet-specific glucose 6-phosphatase related protein
(IGRP) 194. Similarly, screening of a
combinatorial peptide library in a positional scanning format led to the
identification of a peptide derived from dystrophia myotonica kinase (DMK) as
the antigen for the AI4 T-cell clone195. Target of BDC 2.5 (chromagranin)
and BDC 5.2.9 (islet amyloid polypeptide) have similarly been identified.
Although the number of diabetic autoantigens identified in
man and the NOD mouse is expected to continue to increase, currently the
majority of attention is focused on the 6 proteins described in more detail
below.
Preproinsulin
Insulin
and its precursors are obvious target autoantigens in T1D since the hormone is
the major constituent of ß-cells (10-15% of total protein), and indeed
preproinsulin reactive T-cells have been isolated from both diabetic subjects
and NOD mice (reviewed by. Initial studies of islet reactive T-cells from
infiltrates of pre-diabetic NOD mice revealed that insulin reactive clones
predominated 196. Subsequently, the majority were
shown to recognize amino acids 9-23 of the B chain (B:9-23), and when cloned,
were capable of causing accelerated disease in young NOD mice or adoptive
transfer of T1D to NOD.scid animals 197. More recently CD4+
T-cell hybridomas recognizing other epitopes within preproinsulin have been
generated from pre-diabetic NOD mice 198, and spontaneous responses to
proinsulin B24-C33 detected in splenocytes from both pre-diabetic and
newly-diabetic animals 199. Similarly, multiple preproinsulin
epitopes have also been reported in immunized HLA DRB1*0401 transgenic mice 200 and amongst CD4+ T-cells
from the peripheral blood of diabetic patients and their antibody positive 1st
degree relatives 178,
201-204. The relevance of these
observations to diabetogenesis was supported by the recent demonstration that
insulin-specific HLA-DR4-restricted CD4+ T-cells could be expanded
from the pancreatic lymph nodes, but not spleen, of 2 long-term T1D subjects,
but not from those of a DR4+ non-diabetic control 205. Interestingly, TCR recognition
appears to depend upon post-translational formation of a vicinal disulfide bond
between the adjacent cysteine residues in the epitope (A:1-15) 206. The finding that the highly
diabetogenic CD8+ T-cell clone G9C8 recognizes amino acids 15-23 of
the insulin B chain, demonstrated that insulin is also a target for CD8+
T-cells in NOD mice. Recently this has also been shown to be the case in human
T1D 207.
Anti-islet
autoimmunity can be induced in some non-autoimmune prone mouse strains by
immunization with insulin peptides. In particular, administration of B:9-23 to
mice homozygous or heterozygous for H-2d (e.g. Balb/c or Balb/c x
C57BL/6 F1 mice) rapidly induces insulin autoantibodies. Induction is dependent
upon the MHC of the animal, and they are not produced following immunization of
H-2b mice. Surprisingly, the induced insulin autoantibodies cannot
be absorbed with the immunizing B:9-23 peptide, although anti-peptide
antibodies are also produced. It appears likely that the B:9-23 peptide, when
given subcutaneously in saline or incomplete Freund’s adjuvant (IFA), overcomes
T-cell tolerance inducing autoantibodies from what appear to be an existing
population of primed lymphocytes. Consistent with this hypothesis, proinsulin
specific T-cells can be detected in draining pancreatic lymph nodes, but not
the spleen, of unimmunized Balb/c mice. Although the immunized mice develop
insulin autoantibodies they do not develop insulitis, which only occurs with
additional inflammatory signals.
The
ability to induce autoimmunity in non-autoimmune prone animals by peptide immunization
raises important considerations when considering this as a potential
prophylactic therapy. Nonetheless, studies using the NOD mouse have shown that
insulin or B:9-23 given orally 208,
209, subcutaneously, or intranasally 210, prior to the onset of insulitis,
delays the onset and decreases the incidence of diabetes. However, peptide
immunization studies occasionally have unexpected results, as exemplified by
the observation that treatment with the B24-C36 peptide failed due to the
presence of an internal "cryptic" CD8+ T-cell epitope 211. Similarly, whilst intraperitoneal
immunization of 18-day old NOD mice with proinsulin, and subcutaneous
immunization of 5-week old animals with insulin partially protected the
recipients, subcutaneous administration of proinsulin to 5-week old mice
accelerated disease. The mechanism(s) by which tolerance in successfully
treated animals is restored are incompletely understood, but likely involve the
generation of insulin-specific regulatory CD4+ 212 and CD8+ T-cells. One mechanism may relate to the induction of
cytotoxic T cells that kill antigen presenting cells expressing their cognate
peptide 213. It remains to be seen whether the
success in modulating diabetes in the NOD mouse using insulin/proinsulin or
derivatives can be translated to humans. The initial clinical trials of insulin
therapy in pre-diabetic or newly diagnosed diabetic subjects were
disappointing, with no general benefit being observed 214,
215, although some response was
detected in a subset of individuals having the highest titers of insulin
autoantibodies 216,
217 and progression to diabetes of at
risk relatives treated with oral insulin with high levels of insulin autoantibodies
may have been slowed leading to current Trialnet oral insulin trial. However,
there is an issue with the late timing of the interventions in these cases, and
trials at earlier stages in genetically at risk 1st degree relatives
of diabetic individuals who have a single islet autoantibody have been
proposed, although even here the NOD mouse data might suggest that the
autoimmune disease may be too advanced for this form of therapy to be effective
by itself. Use of an altered peptide ligand (APL) of the B:9-23 peptide that
has alanine substitutions at positions 16 and 19 218 has been studied219 with apparently no hypersensitivity
reactions but no benefit.
The
structure of the insulin promotor renders expression primarily ß-cell specific
(reviewed by 220. However, preproinsulin transcripts
and immunoreactivity have also been reported in a minority of thymic and lymph
node cells in both humans and rodents81,
221, 222, likely, at least in part, due to
the action of the autoimmune regulator (AIRE) protein 223. Such expression probably plays a
pivotal role in establishing central tolerance, and may be reduced in
diabetes-prone individuals (reviewed by 224. Thus, expression of particular
variable nucleotide tandem repeat (VNTR) alleles in the human promotor, which
appear to control thymic expression, are associated with disease susceptibility
(IDDM2) 225-227. Similarly, NOD mice lacking
preproinsulin 2, the predominant (if not exclusive) preproinsulin gene
expressed in the thymus of mice 228, show accelerated diabetes, while
thymic over-expression of preproinsulin is protective 229. At present the precise identities
of extra-islet cells expressing proinsulin are a matter of debate (e.g. 230, although recent attention has
mainly focused upon medullary epithelia within Hassall's corpuscles 231-233. Nevertheless, the available data
strongly suggest that the loss of tolerance to (pro)insulin is a critical
risk-factor for T1D, both in NOD mice 69,
70, and humans 205,
225-227, and the establishment of recessive
tolerance to this key molecule can protect from disease 70,
234. Insulin gene expression has also
been reported in multiple organs of diabetic rodents 235, although the significance of this
finding remains unclear.
Immune
responses to insulin, and even to the “same” B:9-23 epitope, can be both
pathogenic and protective. A transgenic mouse with the 2H6 anti-B:9-23 T-cell
receptor prevents diabetes dependent upon TGFß production. A TCR transgenic
(BDC 12-4.1) and retrogenic (BDC 12-4.4) cause disease 236 (Note 12-4.4 sequence variants
differ in pathogenicity). Of note, most of the T-cell receptors recognizing
insulin B:9-23 utilize a conserved VaT-cell receptor segment
(TRAVSD-4*04). These alpha chain T-cell receptors can pair with multiple very
different TCR beta chains and recognize the B:9-23 peptide. In addition, the N
and V region alpha chain sequences targeting the B:9-23 peptide are variable.
This has led to the hypothesis that propensity for insulin autocomponents may
be genonomically determined by common Vachain sequences targeting an
invariant insulin peptide sequence (B:12-22) 237. Genetically determined
abnormalities of tolerance maintenance and environmental activation might act
upon an immune system poised to target insulin.
There
has been progress in defining human T-cells targeting proinsulin and insulin.
Kent and coworkers detected DR4 restricted clonally expanded pancreatic lymph
node T-cells recognizing insulin A1-15 peptide. Peakman defined CD8 T-cells
recognizing epitopes from the human preproinsulin leader sequence presented by
HCA2 (PPI17-24 WGPDPAAA and PPI15-24 ALWGPDPAAA) 141. By Y-interferon ELISPOT,
approximately 50% of new onset patients had circulating CD8+ T-cells reacting
with these peptides. Reactivity was greater than to other HLA-A2 studied
proinsulin peptides (B10-18; B18-27; C22-30; C27-35; C31-A5; and A1-10). Three
clones from a single patient all had same TCR ß chain TRBV12-4 (two achains
TRAV 12-3 and TRAV 13-2). The clones recognizing PPI15-24 showed
enhanced cytotoxicity when islets were cultured in high glucose.
Durinovic-Bello identified a CD4 DRB1*0401 T-cell epitope (proinsulin 73-90)
with core sequence LALEGSLQK (human cone 52c1)238. T-cells reacting with this epitope
upon in vitro expansion can be isolated utilizing a DRB1*0401 tetramer. In
evaluation of a series of proinsulin peptides (e.g. C19-A3 GSLQPLALEGSLQKRGIV)
presented by DRB1*0401, Arif and coworkers find responses of both patients and
controls but patient responses are proinflammatory (IFN-γ) while on
ELISPOT controls produce IL-10 to some peptides.
Work
by Stadinski et al. brought a new focus to the details of antigen presentation
in type 1 diabetes 239. This study used a method of
register trapping to determine which “face” of B:9-23 epitope is recognized by
T cells, and which binds to I-Ag7. The register in which a peptide
binds to MHC is defined as the specific linear orientation of the peptide
within the MHC groove. As one moves a
peptide one amino acid up or down the groove of the MHC, the peptide has to
rotate to bind to the pockets of the MHC,
changing the side chains facing outward and interacting with T cell
receptors. By fixing the way the insulin B:9-23 peptide binds to MHC II, Stadinski and coworkers showed
that B:9-23 stimulated a panel of insulin-specific CD4+ T cell hybridomas only
when bound in “register 3” (SHLVERLYLVCGEEG; the
downwardly pointing residues are mutated anchor residues for p1 and p9 pockets
of I-Ag7). This directly
contradicted findings by Levisetti et al. who proposed register 1 and 2 as
being recognized by the two classes of T cell hybridomas 240. Levisetti measured T cell
responses to N- and C-terminally deleted versions of B:9-23, and interpreted
diminished IL-2 production as
elimination of B:9-23 binding in a specific register. However, changes in T
cell responses could have also been explained as elimination of an important TCR
epitope(s) instead.
Another
paper from Kappler’s group showed that some truncations of the B:9-23 peptide
might in fact increase T cell reactivity. For example, C-terminal truncation to
B21 eliminates the arginine residue that conflicts with the I-Ag7 p9 binding pocket when bound in register 3, and truncation
to B20 eliminates glutamic acid that interferes with recognition by some T cell
clones241.
Building on this knowledge, the group described generation of a series
of B:9-23/ I-Ag7 tetramers that stained majority of insulin-specific
CD4+ T cell hybridomas. These tetramers displayed slightly modified B:9-23
epitopes bound in register 3 to I-Ag7. In addition to the
hybridomas, the B:9-23 tetramers could also stain about 5% of primary CD4+ T
cells present in the pancreas of NOD mice. The authors also proposed a model
explaining how the “register 3” peptide might be recognized as “de novo”
antigen expressed specifically in the pancreas. According to this model,
pancreatic ß cells generate truncated version(s) of the peptide that are not
present in the thymus during maturation of thymocytes. Absent during negative
selection, this peptide(s) cannot be used to delete autoreactive thymocytes
recognizing insulin. Thus, this unique version of the peptide can be considered
as “neo-antigen”. An ability of
pancreatic ß cells to process insulin has been previously described by Mohan et
al. 242. This paper showed that small
percentage of ß cells had secretory granules containing B:9-23 peptide, and
that this peptide could be “picked up” by resident dendritic cells. Therefore,
it is conceivable that this unique “register 3” insulin peptide can be
generated only in the pancreas.
Additional support for “register 3” recognition
came recently from the lab of Harald von Boehmer 243. In this study, Daniel et al. used
an insulin B:9-23 register 3 binding mimetope to prevent diabetes in NOD mice.
They slowly infused low levels of the mimetope, and showed that this regiment
generated insulin-specific regulatory T cells. They also showed that such
treatment induced dominant tolerance since polyclonal responses to insulin were
blocked.
The insulin peptide B:9-23 can be
targeted by T cells not only in mice with I-Ag7 but also by I-Ab
in a model created by the laboratory of Massimo Trucco. In this model (ID-
TEC: insulin deleted thymic epithelial
cell) an insulin 2 knockout mouse is combined with Aire driven Cre insulin 1-/-
deleting insulin 1 in thymic epithelial cells. These C57 mice with I-Ab rapidly
develop diabetes and have large numbers of T cells producing interferon gamma
in response to stimulation by insulin or the B:9-23 peptide244.
In summary, identification of
register 3 presentation of B:9-23 peptide opened new avenues of research.
First, the panel of I-Ag7/B9-23 tetramers will enable in vivo
studying of insulin-specific, I-Ag7 restricted CD+4 T cells in NOD
mice. Second, the analogous approach
might lead to generation of similar reagents for human system. Finally,
Daniel’s study shows that administration of insulin peptide mimetope designed
to bind in relevant register can induce dominant tolerance that prevents
autoimmune activation of T cells in the pancreas245.
Glutamic Acid Decarboxylase
Like insulin, glutamic acid decarboxylase (GAD) is a major autoantibody target
in human diabetic patients (reviewed by 246.
It catalyzes the formation of the inhibitory neurotransmitter aamino-butyric
acid (GABA), and is expressed on synaptic vesicles in many regions of the
central nervous system and multiple neuroendocrine tissues. GAD exists in two non-allelic
forms, GAD65 and GAD67, which are 65% identical, differing primarily in their
initial 250 residues, but the precise distributions of the isoforms differ
between man and rodents. Thus, GAD65 is the major form expressed in the human
pancreas, where it is predominantly localized to ß-cells, whereas GAD67 is the
major isoform expressed by mouse islets, albeit at barely detectable levels 247,
248. Such differences provide important
considerations for extrapolating data regarding GAD obtained from mouse models
to the human disease.
Unlike
humans, spontaneous antibody responses to GAD are rarely, if ever, detected in
NOD mice 248,
249, although T-cell responses are
amongst the first autoreactivities detected in neonatal females, being apparent
as early as 3-4 weeks of age 250,
251. Initial responses to GAD in NOD
mice are directed to the fragment GAD65 (509-543), which contains at least two
overlapping I-Ag7 restricted determinants (residues 524-538 and
530-543), each eliciting T-cells of distinct phenotypes and showing particular
TCR Vß gene usage 252. Thus, spontaneous GAD-reactive CD4+
T-cells from young NOD mice primarily recognize the immunodominant epitope
530-543 (p530). However, T-cells to the overlapping determinant 524-538 (p524)
dominate the response after immunization with GAD65 (524-543). p530-responsive
T-cells typically use the Vß4 gene, whereas the Vß12 gene is preferentially
used to encode the TCR of p524-responsive T-cells. The p524 responsive T-cells
appear to be regulatory and upon adoptive transfer to young NOD mice can
inhibit diabetes development. During the course of disease determinant
spreading occurs generating T-cells recognizing additional epitopes towards the
N-terminus of GAD65. Similarly, immunization with recombinant GAD65 or GAD67
reveals additional epitopes 253,
254, some of which are shared between
the two isoforms. GAD-reactive CD8+ T-cells have also been
identified 255.
GAD65
is also a major target of T-cell autoimmunity in human T1D, with peripheral
blood mononuclear cells (PBMCs) from approximately 50% of new onset patients
responding to this autoantigen Atkinson 256. HLA-DR4:GAD65555-567
reactive CD4+ T-cells can also be detected in the peripheral blood
of healthy subjects, although in contrast to diabetic individuals these cells
are primarily restricted to the naive pool 257,
258. Intriguingly, in a cohort of
at-risk 1st degree relatives there was an inverse relationship
between cellular and humoral autoreactivity to GAD65. The significance of this
observation remains uncertain, but it may reflect the fact that binding of some
GAD65-specific antibodies to their target can suppress presentation of certain
immunodominant T-cell epitopes 259, although the converse has also
been reported 260. A number of CD4+
epitopes within human GAD65 have been mapped using immunized transgenic mice 261 or T-cells isolated from newly
diabetic subjects and at-risk relatives 262-269.
These studies have provided evidence for molecular mimicry to both
exogenous and endogenous antigens. Thus, cross-reactivity of T-cells from
recent onset HLADR3/4 positive patients between rubella virus envelope protein
1(157-176), RVE2 (87-107) and GAD65 (274-286) was observed 263. Similarly, a GAD65 (339-342)
restricted T-cell clone from a pre-diabetic patient also responded to residues
674-687 of human cytomegalovirus major DNA-binding protein of 134kDa 270, an observation that appears
especially relevant given the reported association of cytomegalovirus infection
and T1D 271. Other apparent cross-reactivities
that have been reported include GAD65 (247-279) with Coxsackie virus B3 P2-C
protein (32-47) 272,
and GAD (506-518) and proinsulin (24-36) 273, although these conclusions are
disputed by a more recent study using cloned T-cells 274. As in the NOD mouse, GAD-reactive
CD8+ T-cells have also been identified in human T1D patients 275.
Like
other autoantigens, the precise role of GAD65 in the onset and progression of
spontaneous T1D in humans and NOD mice remains uncertain 276. Thus, in one of three transgenic
lines of NOD mice expressing an antisense construct shown to reduce the
expression of both GAD65 and GAD67, not only was diabetes and insulitis
abolished, but also T-cells derived from these mice were unable to transfer
disease 277; a result which suggests that GAD65
plays a critical role in T1D development. There has been no follow up of this
study and it is possible that disease prevention related to introduction of
chromosomal region from knockout rather than effect of GAD knockout.
Conflicting results have come from other studies. For example, NOD mice
homozygous for a disrupted GAD65 gene still develop diabetes, and NOD mice
rendered tolerant to GAD by transgenic expression of a modified form of the
protein under control of the invariant chain promotor, still develop T1D with
normal incidence 278. The reasons for the contradictory
outcomes are unclear, and have been the subject of debate (e.g. 277), but it seems likely that the
protection afforded by the antisense construct is not solely due to the
immunological effects of suppression of GAD, and that GAD65 is unlikely to be
essential for diabetes development in NOD mice, although given the differences
in expression, this conclusion cannot be extended to humans.
Nonetheless,
GAD65 is a legitimate target for immunotherapy, and administration of GAD65
intrathymically, orally, nasally or in the form of epitope peptides to 3-week
old NOD mice protects against diabetes 250,
251, 279-282. Intramuscular injection of a
recombinant adeno associated virus expressing GAD500-585 to 7-week
old female NOD mice also induced tolerance 283. Similarly, phase 2 clinical trials of new onset type 1
patients classified as latent autoimmune diabetes of the adult with
alum-formulated recombinant GAD65 demonstrated preservation C-peptide levels in
the treated subjects at 24 weeks post-therapy. GAD67, though not a major
diabetes autoantigen may also protect against disease 284, and GAD65 expressed at high levels
in pancreatic ß-cells under control of the rat insulin promoter was protective
in one of two transgenic lines 285, likely due to an increase in the
proportion of IL-10 producing T-cells. In addition, the islets from NOD.scid transgenic RIP-GAD65 mice were
partially protected from adoptive transfer of diabetic splenocytes. In
contrast, widespread expression of GAD65 under control of the MHC class I
promoter did not induce tolerance in NOD mice 286, and in one of three transgenic
lines disease was exacerbated with a greater degree of insulitis in males and
an increased overall incidence of T1D. Transgenic and retrogenic expression of
T-cell receptors targeting GAD do not induce diabetes 287 but can protect NOD mice 288 and a human TCR transgenic
developed insulitis 289. A recent study of anti-GAD
retrogenics by Vignali and coworkers suggests that there is insufficient GAD in
islets of mice to allow T cell
targeting, with the anti-GAD retrogenic mice dying of encephalitis with
no evidence of insulitis despite induction of GAD autoantibodies 61.
A large trial of GAD in alum immunization failed to alter loss of
immunization c-peptide of new onset patients290.
.
IA-2
and phogrin
In addition to proinsulin and GAD65, another major autoantibody targets in
human diabetic subjects defined at the molecular level are the protein tyrosine
phosphatase (PTP) superfamily members IA-2 (also known as ICA512 and PTPRN) and
phogrin (also known as IA-2ß, IAR, ICAAR, PTPRP, and PTPRN2) (reviewed in 291)292. Approximately 60-70% of new-onset
T1D patients react to both molecules, as does 40-50% of 1st degree
relatives 293-299, but less than 2% of controls. In
multiple antibody positive individuals IA-2 is typically one of the last to
become evident, and consequently is highly predictive for progression to disease
300,
301. Initially described as
autoantibody-reactive 40 and 37 kDa tryptic fragments derived from non-GAD 64
kDa precursors immunoprecipitated from radio labeled ß-cells 293,
298, 302, they were subsequently shown to be
proteins that had previously been cloned independently by several groups using
subtractive and expression screening strategies 294,
299, 303-310.
Like the GAD isoforms, IA-2 and phogrin show over-lapping
but distinct expression patterns in the brain and multiple neuroendocrine cell
types, but are expressed at much higher levels than GAD in islets, and are
localized to dense core secretory granules rather than synaptic vesicles 305,
311. In addition, they each exist in a
number of splice forms, which are differentially expressed in various tissues 312, JC Hutton, personal
communication). Both IA-2 (979 amino acids, encoded on human chromosome
2q35-36.1) and phogrin (986 amino acids, encoded on human chromosome 7q36) are
type 1 integral membrane glycoproteins, and have cytoplasmic domains (378 and
375 amino acids respectively) that are ~74% identical 291. In contrast there is only 27%
indentify within their extracellular domains.
All humoral responses so far identified map to the cytoplasmic domains 296,
313, 314, and 90% of IA-2 reactive sera also
detect phogrin, whilst 99% of phogrin-reactive sera detect IA-2, yet other PTP
family members are not targeted 315. Initially synthesized as ~130kDa
glycosylated precursors, IA-2 and phogrin are cleaved at a consensus dibasic
cleavage site in the late Golgi/granule to produce the mature 62-64kDa proteins
305,
316. Following glucose-stimulated
insulin secretion IA-2, but not phogrin, is cleaved by µ-calpain within its
cytoplasmic domain 317.
At present the precise functions of IA-2 and phogrin within
pancreatic ß-cells remain uncertain. The orientation of the mature proteins
in the granule membrane (NH2 terminal ~200aa in the lumen/ ~400aa
COOH terminal PTP domain in the cytoplasm) suggests possible roles as receptors
that signal to the cytoplasm from either the granule lumen or the extracellular
environment, however the nature of the presumptive ligands, and the signal
transduction mechanisms, are unclear 311,
318. Changes in sequence surrounding
the consensus active site in their PTP domains are predicted to preclude
catalytic activity, and the proteins have little, if any, phosphatase activity
towards common substrates 305. Nevertheless, they adopt a similar
fold to active PTPs (JC Hutton, personal communication), and activity can be
restored in either IA-2 isoform by mutagenesis of 2-3 amino acids 319. Given that several other
receptor-type PTPs contain tandem PTP domains, one of which is active and the
other inactive and likely regulatory 292 it has been proposed that IA-2 and
phogrin may interact with an as yet unidentified active PTP, but at present
this remains conjecture. Regulated interaction of IA-2 with the PDZ domains of
ß2-syntrophin, and nNOS has been demonstrated in rat insulinoma cells 317,
320, but to date no interaction
partners have been defined for phogrin. Nonetheless disruption of either the
IA-2 or phogrin genes in mice produces a similar phenotype; mild glucose
intolerance due to impaired insulin secretion 72,
321.
Unlike
humans, NOD mice produce circulating antibodies to IA-2/phogrin at relatively
low frequency, and they do not show disease specificity 322. In contrast, T-cell responses to
both autoantigens have been detected in both murine and human T1D 301,
323-326. To date attention has mainly
focused upon CD4+ responses to the conserved cytoplasmic domains,
although a potential epitope in the lumenal domain of rat IA-2 was recently
identified by pooled sequencing of peptides eluted from rat insulinoma cells
transfected with cDNAs encoding HLA-DR4 327. Similar to studies of proinsulin
by ELISPOT assays, patients with type 1 diabetes produce DRB1*0401 restricted
proinflammatory responses to IA-2 (e.g. 652-80; 709-35; 752-75; 793-817;
853-72; 955-76) while controls respond but primarily with IL10 production.
Thus, immunization of NOD mice with recombinant rat phogrin cytoplasmic domain
in CFA induced CD4+ Th1 biased responses focused upon two major
epitopes, amino acids 640-659, and amino acids 755-777 328. Subsequently, spontaneous
proliferative responses to residues 755-777, but not 640-659, were detected in
splenocytes and lymph nodes from young prediabetic NOD mice 329. No responses were observed in
splenocytes from Balb/c mice of the same age. Although none of the cloned
phogrin-reactive T-cells accelerated disease when transferred to prediabetic
NOD mice, some were able to destroy transplanted rat islets and cause diabetes
in a NOD diabetes recurrence model 325.
The phogrin 640-659 and 755-777 peptides also induced
responses in immunized HLA-DQ8 transgenic mice, and the same specificities were
detected in 17% and 35% respectively of ICA positive prediabetic individuals,
but less than 5% of age- and HLA-matched controls 330. Surprisingly, proliferative
responses to the phogrin antigenic peptides were seen in PBMCs from some
HLA-DQ8 negative individuals, suggesting that they might be “super-epitopes”
recognized by multiple HLA-DR and –DQ molecules.
The cross-reactivity between IA-2 and phogrin autoantibodies
and extensive sequence conservation, suggests that T-cells recognizing both PTP
isoforms might also be generated. Initial studies did not support this
hypothesis; none of the T-cell clones isolated from NOD mice immunized with the
cytoplasmic domain of phogrin recognized epitopes derived from IA-2, and
similarly, T-cells from IA-2 C-terminus immunized mice did not react to
processed phogrin (K Kelemen personal communication). Consistent with this
observation, the 2 immunodominant epitopes presented by I-Ag7 from
processed IA-2 (residues 685–701 and 725-741; JC Hutton and DAA Vignali
personal communication) are not conserved between the 2 isoforms. Peptides containing these epitopes, which are
100% conserved between mouse and human IA-2, also induced responses when used
to immunize NOD mice 331, although the highest stimulation
index (SI) was obtained following administration of the peptide linking these
two epitopes which is conserved between IA-2 and phogrin, but was not detected
in the initial studies using naturally processed antigen. However, more
recently a T-cell hybridoma generated from NOD mice immunized with phogrin was
shown to recognize a cross-reactive epitope of IA-2 (HW Davidson, unpublished
observation) raising the possibility that this might also occur in human
diabetic subjects. Like phogrin, spontaneous responses to IA-2 have been
detected in NOD mice, although in contrast to phogrin, IA-2 specific T-cells responded
by cytokine secretion but not proliferation 329,
332. The epitope(s) spontaneously
recognized have yet to be defined.
Spontaneous responses to IA-2 C-terminal epitopes have also
been observed amongst 326,
333, 334, 334. For example, 6 sets of peptides
nested around defined core sequences were eluted from HLA-DR4 molecules
expressed by B lymphoblastoid cells that had processed recombinant protein. At
least one of the peptides corresponding to these epitopes produced
proliferative responses (SI>2) in PBMCs from 9/13 HLA-DR4 positive diabetic
patients, but 0/8 –DR4 positive non-diabetic controls 334. Similarly, Elispot analyses conducted using a peptide library from the
IA-2 cytoplasmic domain revealed a 17-fold increased frequency of IFNγ
positive cells in PBMCs from DR4/DQ8 positive T1D subjects as compared to
matched controls 326.
CD8+ T-cell responses to IA-2 have also been
detected, although these appeared not to be disease specific 335. In this regard it should be noted
that the phosphatase domains of phogrin and IA-2 show high evolutionary
conservation with homologs in zebrafish (~80% identity), Drosophila (FLYDA; ~60% identity) and C. elegans (B0244.2 gene product IDA-1; ~50% identity) 336,
337, which are all greater than the
conservation of IA-2 with other mammalian PTP domains (<40% identity) 292.
This suggests that exposure to common multi-cellular pathogens could
trigger cross-reactive T-cells through molecular mimicry, although this
hypothesis remains to be established. Molecular mimicry with multiple viruses
including rotavirus, Dengue, cytomegalovirus, measles, hepatitis C,
rhinoviruses, hantaviruses, and flaviviruses, as well as the bacterium
Haemophilus influenza, and several milk, wheat, and bean proteins has also been
proposed 333, although likewise direct evidence
to support this suggestion is currently lacking.
Like GAD65, neither IA-2 nor phogrin is essential for T1D
induction in NOD mice 72,
73, whilst attempts to target the
molecules therapeutically have given mixed results. Thus, immunization of 3-4
week old NOD mice with phogrin peptide 755-777 in saline reduced the incidence
of T1D (JC Hutton, personal communication), but administration of recombinant
IA-2 in IFA exacerbated disease 332. Thymic expression of IA-2 has been
documented 312,
338, and in common with other
tissue-specific antigens, involves cells of the medullary epithelium. This suggests that most IA-2
restricted T-cells are likely to be subject to negative selection. However, the
major splice-variant of IA-2 expressed in the thymus bears a deletion in exon
13 312, which could allow autoreactive
cells directed to this region to escape negative selection. Interestingly, the
exon 13 deleted variant, which also is expressed in pancreatic islets 312,, lacks the transmembrane domain,
and as a consequence the carboxy-terminal domain is predicted to enter the lumen
of the endoplasmic reticulum. This might allow a potential site of N-linked
oligosaccharide addition to be used (aa766-768), significantly affecting the
processing of the molecule by APCs.
IGRP
First identified as a ß-cell specific protein in a subtractive
hybridization screen that was performed to identify ß-cell specific proteins
that could be autoantigens, or regulators of insulin stimulus-secretion
coupling, IGRP was recently
identified as the target of the diabetogenic NY8.3 clone. It is expressed in a highly
pancreatic ß-cell specific manner 339 yet appears to be controlled by a
different set of transcription factors than those regulating other ß-cell genes
such as insulin and amylin 340. Due to its sequence homology with
glucose 6-phosphatase (G6Pase) it has been investigated as a potential
component of a glucose substrate cycle potentially controlling energy
metabolism in the ß-cell 341, however to date no catalytic
activity has been demonstrated 342. The human IGRP gene is located on
chromosome 2q 24-31, a short distance from the glucagon and GAD67 genes, in a
region where IDDM7 and NIDDM and the Bardet Biedl genes map. Like the liver
G6Pase, IGRP bears a carboxy-terminal KKXX sequence typical of endoplasmic reticulum
resident transmembrane proteins, and most of its sequence appears buried in the
membrane with only short cytoplasmic and lumenal peptide loops 341-343.
IGRP mRNA is abundant in mouse and human islets, but it is a
pseudo-gene in the rat. Humoral autoreactivity to IGRP has been
searched for in NOD mice and human diabetic subjects without success (JC
Hutton, personal communication).
The
NY8.3 CD8+ T-cell clone was isolated over 10 years ago by Yoon and
colleagues from islet infiltrates of acutely diabetic NOD/Lt mice. It is
restricted to H2-Kd, and in the presence, but not absence, of CD4+
T-cells can transfer disease to irradiated NOD mice. Circumstantial evidence
suggests that the target of NY8.3 may be associated with the initial insulitic
response. Thus, CD8+ T-cells with closely related, or even identical,
a chains to NY8.3 (Va17 and Ja42
elements joined by the N-region sequence MR(D/E)), but distinct ß chains, are
prevalent in NOD insulitic lesions as early as 4-5 weeks of age 344, whilst NOD mice transgenic for the
NY8.3 ß chain (Vß8.1) show accelerated T1D, and the majority of infiltrating
CTLs from diabetic animals express an endogenous a chain identical to that of NY8.3 345. Disease onset is further
accelerated in NOD mice transgenic for both NY8.3 TCR chains in both male and female
animals, although overall incidence is unchanged 346. NY8.3 TCR transgenic animals on a
NOD/RAG2-/- background develop diabetes less frequently and at a
slower rate than RAG2+ 8.3-NOD animals (albeit to a much greater
extent than non-transgenic NOD/RAG2-/- animals which are protected
from T1D), but rapidly progress to disease following adoptive transfer of
non-transgenic CD4+ T-cells 346, further confirming the importance
of CD4+ cells in the recruitment of naïve CD8+ cells to
islets. Despite the high frequency of CD8 T-cells targeting IGRP, induction of
tolerance to the molecule does not influence progression to diabetes 76,
77 and even the TCR anti-IGRP
transgenic requires immune response to insulin for spontaneous diabetes. Nevertheless, peptides of IGRP can
be used to prevent the development of diabetes of NOD mice 347.
A
mimotope was defined for the NY8.3 TCR (NRP), and subsequent cytotoxicity
assays indicated that almost 50% of the CD8+ T-cells from islet
infiltrates of non-transgenic animals recognized this peptide 348. Similarly, H2-Kd
tetramers bearing an NRP variant showing higher agonistic properties (NRP-V7) 349, stain up to 37% of the CD8+
positive T-cells present in NOD islet infiltrates from 11-14 week old animals,
and a significant percentage in the circulation 350. More recently, H2-Kd-NRP-V7
coated superparamagnetic nanobeads have been used to selectively label NY8.3
TCR transgenic T-cells, and their recruitment to islets studied in real-time by
magnetic resonance imaging (385)351. Tetramers bearing residues 206-214
of mouse IGRP, the epitope identified by sequencing of peptides from NIT1
insulinoma cells, similarly stain a significant proportion of islet infiltrating
cells (JC Hutton, personal communication). Interestingly, studies using NRP and
its derivatives demonstrated a time-dependent increase in the functional
avidity of the peripheral clonotypic response that may contribute to the
progression from benign to destructive insulitis. Subsequent analyses indicated
that the observed avidity maturation correlated with differential usage of 3
distinct Va17 elements that created TCRs with differing binding
affinities for the natural ligand. Thus, at 9 weeks a significant population of
low affinity Va17.6 expressing cells were present,
whilst by 20 weeks these had been replaced by high affinity Va17.5
expressing clonotypes 352, presumably reflecting differential
peripheral expansion of the high avidity clones at the expense of their low
avidity counter-parts. Interestingly, some altered peptide ligands targeting
IGRP206-214 specific T-cells could prevent T1D, but only under
conditions that spared low avidity clonotypes; higher doses or affinities that
caused virtually complete ablation of this response led to increased
recruitment of alternative, sub-dominant specificities to islets and disease
progression 353. There is evidence for targeting of
IGRP by patients 354.
ZnT8
Autoantibodies
to the islet zinc transporter ZnT8 are prominent in man but apparently absent
in NOD mice 185,
355-358.
The ZnT8 molecule associated with beta cell secretory granules
transports zinc from the cytoplasm to the granule where it complexes with
insulin forming insulin crystals. Multiple epitopes of ZnT8 are recognized by
autoantibodies including a polymorphic amino acid associated with Type 2
diabetes risk 185. Patients who target only the
arginine variant of ZnT8 are homozygous for the arginine polymorphism, while
those targeting the tryptophan variant are homozygous for the tryptophan
variant of molecule 359.
This confirms the autoimmune nature of targeting self in Type 1 diabetes360.
ZnT8 autoantibodies disappear most rapidly after onset of diabetes. In
man, multiple different peptides are recognized by T lymphocytes186.
Chromogranin A
Haskins
and coworkers have recently identified chromogranin A 53 as the target of the classic T cell
BDC 2.5 clone that has been utilized to study the pathophysiology of islet
autoimmune for more than 20 years 56,
169, 361.
This neuroendocrine molecule is widely expressed and is processed to
yield multiple peptides. To date, no
autoantibodies reacting with chromogranin A are described. Given its widespread
neuroendocrine distribution, it is remarkable that tissue destruction by the
BDC 2.5 clone appears to be islet specific. As reported, the recognition by the
BDC 2.5 T cell receptor is very specific reacting with a specific
neuroendocrine produced peptide of chromogranin (WE-14) such that the peptide
does not fill all of the I-Ag7 groove and a relatively long c-terminal
extension of the peptide projects from the MHC groove 53.
Adding the native amino acids at the N-terminus to the groove or
removing c-terminus amino acids of the EL-14 peptide abrogates BDC 2.5 reactivity. Thus it appears that only tissue specific
processing of chromogranin can create the relevant recognized sequence.
Molecular recognition of target
autoantigens
A
developing consensus is that the peptide determinants driving autoimmune
disorders may be recognized by their cognate T cell receptor in unusual
conformations such as not completely filling the MHC groove 53, in low affinity MHC binding
registers 239, with T cell receptors bound at
unusual angles 362 and with post-translational
modifications 363. In addition, tissue specific
peptides may be generated in the specific target cell in a manner unlikely to
be recapitulated in antigen presenting cells or thymic epithelial cells 53,
242.
All the above properties suggest mechanisms that can contribute to
autoreactive T cells escaping negative thymic selection. The processing of chromogranin A to the
specific peptide recognized by the BDC2.5 T cell receptor is a prominent
example of only partially filling the groove of I-Ag7 and requiring specific
neuroendocrine processing 53.
The B:9-23 peptide studied by Unanue and coworkers and Keppler and
coworkers is another example 242.
ICA69
Originally identified by an expression cloning approach using
sera from pre-diabetic ICA positive individuals who subsequently progressed to
overt disease 364, the product of the ICA1 gene was
recently shown to be an arfaptin-related protein associated with the Golgi
apparatus 365. Like GAD65 and IA-2, ICA69 is
expressed in multiple neuronal and endocrine cell types, but may also be
expressed in other peripheral tissues including the heart and exocrine pancreas
366,
367, and antibodies reacting with ICA69
are found in patients suffering from several other immune disorders besides
T1D. To date no group has established a specific ICA69 auto-antibody assay to
assess their predictive power in pre-diabetic subjects. Tissue-specific expression
is controlled by alternative core promotors 368, and in NOD mice, thymic expression
of ICA69 is greatly decreased relative to non-autoimmune prone strains 369, although the significance of this
is unclear. Disruption of the ICA1 gene does not protect NOD mice from
spontaneous T1D, but intriguingly ICA69null animals resist disease acceleration
by cyclophosphamide 370.
T-cell responses to ICA69 have been detected in diabetic
subjects 371,
372 and NOD mice 373. Interestingly, the immunodominant
epitope of ICA69 (residues 36-47) shows immunological cross-reactivity with the
ABBOS epitope in BSA 374, which together with
epidemiological studies, has prompted speculation that BSA from cow’s milk
might be a triggering antigen in human T1D 375. However, responses to ABBOS are
observed in PBMCs from both diabetic subjects and healthy controls 376, casting doubt on this suggestion.
Interestingly, T-cell responses to ICA69 in diabetic subjects showed a positive
correlation with HLA-DR3, but inverse relationship with antibody positivity 371, and increased tendency to anergy
relative to other T1D autoantigens 377.
Carboxypeptidase E
Carboxypeptidase E (CPE) is a neuroendocrine specific
prohormone processing enzyme that selectively removes carboxy-terminal basic
residues from precursor proteins and peptides (reviewed by 378,
379. Also known as carboxypeptidase H,
CPE is a major component of insulin secretory granules 380, and is co-secreted with insulin by
pancreatic ß-cells 381.
It was shown to be a serological autoantigen in man, but is not a
humoral autoantigen in NOD mice. Presumptive T-cell responses have not been
defined, although a recent study using HLA-DR4 transfected INS1 cells
identified potential epitopes within the CPE molecule 327.
Amylin
Also known as islet amyloid polypeptide (IAPP), amylin is a
secretory granule protein localized primarily in pancreatic ß-cells, but which is also expressed to a limited
extent in gastric endocrine tissue
(reviewed by 382. Like insulin it is a
disulfide-linked heterodimer that is initially synthesized as a single-chain
precursor 383,
384, from which the mature protein is
excised by prohormone convertases 385. At present the precise
physiological role(s) of amylin are uncertain, although it is probably involved
in controlling food intake and body weight 382. The human protein is capable of
forming amyloid fibrils, and has been implicated in the pathogenesis of type 2
diabetes. Recently, an HLA A*0201 restricted epitope within the signal sequence
of amylin, which is cleaved in the endoplasmic reticulum, was identified using
PBMCs from human diabetic subjects 386.
The initial indication that IAPP might play a role in NOD
mice came from mapping studies showing genetic linkage between IAPP locus and a
candidate antigen for the BDC6.9 NOD CD4+ T-cell clone 387. The same group reported that
islets from NOD.IAPP-deficient mice could not stimulate BDC6.9 clone, but the
authors failed to identify the relevant stimulatory peptide 52. However, the same paper presented
convincing evidence that another clone, BDC 5.2.9, recognizes KS20 peptide
derived from the sequence located between propeptide 1 and propeptide 2 of IAPP
protein. Given the highly diabetogenic nature of BDC5.2.9 clone, and the data
mentioned above, one might conclude that IAPP serves as an autoantigen in NOD
mice, and that it might contribute to the human disease.
Hsp 65
Heat shock protein of 65kD (Hsp65), now commonly referred to
as Hsp60, is a member of the broadly expressed, and evolutionarily highly
conserved, HSP60/GroEL/chaperonin family. Although most of the research
relating to this protein has focused upon its role in protein folding, it has
recently attracted considerable interest as a potential stress cytokine283,
284, and has been implicated in several
inflammatory autoimmune diseases388. In mammals the HSP60 family
comprises cytosolic and mitochondrial isoforms. The cytosolic form is released
from stressed cells 286 and during necrosis 287, and may also be a component of
secretory granules in neuroendocrine cells389. The precise role of secreted Hsp60
in innate immunity is currently a matter of controversy, with reports of
pro-inflammatory properties mediated through CD14, Toll-like receptor 2 (TLR2)
or TLR4 (e.g. 289 being recently ascribed to
contamination with TLR ligands such as LPS390. A primarily anti-inflammatory role
has also been proposed391.
Bacterial Hsp60 is highly immunogenic, and induces
antibodies that cross-react with mammalian Hsp60 and are pro-inflammatory.
However, in pathogen-free NOD mice spontaneous T-cell responses to Hsp60 that
precede detectable humoral responses to this antigen, can be observed prior to
the onset of insulitis392,
393. Hsp65-specific T-cells transfer
diabetes to young NOD mice 392, and immunization of young animals
with the antigen in IFA triggers disease, although this effect is transient and
the mice show long term protection 392. In contrast, immunization of some,
but not all, non-autoimmune prone strains of mice with residues 437-460 of
Hsp65 (p277; 394) conjugated to ovalbumin or BSA
induces a non-remitting T1D which could be transferred to naïve animals by
p277-specific T-cell lines 291. Protection from spontaneous T1D is
provided by immunization of young NOD mice with the protein in PBS392, and of cyclophosphamide
accelerated T1D by DNA vaccination with Hsp65 cDNA 292. Peptide immunization may also
reverse established disease in NOD mice 395, although this result has been
disputed 396. Moreover, treatment of donor
animals with p277 prevents adoptive transfer of disease to NOD.scid animals by T-cells from islet
infiltrates 397, and arrests streptozotocin-induced
disease 398, likely due to the induction of a
Th2 biased response 296,
297. However, intra-thymic
administration of p277 did not protect NOD mice from spontaneous disease, but
tended to exacerbate it 298. In addition, a study by Bowman and
colleagues failed to replicate studies in NOD mice showing protection from
diabetes with P277 peptide 396. Surprisingly, administration of
the Th1 p277-restricted clone C3.5 protected NOD mice from spontaneous disease 399, presumably due to attenuation of
the clone as was previously reported for the related C9 line 400.
Hsp65 is also a T-cell autoantigen in humans 401-403 although it is uncertain if this
response is disease-specific. Interestingly, although no correlation between
IFNγ producing Hsp65 specific T-cells and diabetes risk was observed in
discordant monozygotic twins, low-risk (ICA negative) individuals showed
significantly enhanced IL-10 producing Hsp65 specific T-cells 300. On the basis of the protection
afforded to NOD mice 395 a limited phase II clinical trial
with a humanized version (DiaPep277) has been conducted in new onset adult
patients. Interestingly, this showed preservation of C-peptide levels for at
least 10 months, with reduced exogenous insulin requirement, in the treated
individuals404. Further phase II and III trials
were conducted with inconclusive results 405,
406.
Primacy and diversity of the molecular targets of T-cells in
diabetes
As discussed above, the concept that diabetic autoimmunity
in humans is initiated by T-cell mediated attack directed at a single ß-cell
specific molecule, while attractive is as yet unproven though with considerable
evidence that insulin/proinsulin may be such a primary target. That this can be
the case is clearly demonstrated by the various mouse models in which
neo-antigens are expressed under control of the RIP (for example 80,
96, 97 or where activated diabetogenic
T-cell clones are transferred to susceptible lymphopenic animals (for example51,
308. However, it is uncertain if it is
the case in the natural disease, where the triggering event remains
ill-defined, although it does appear likely that the initial response targets
only a limited number of autoantigens. In NOD mice there is a restricted islet-reactive
TCR repertoire in early insulitic lesions although surprisingly, neither GAD65
nor insulin-specific clones predominate 173,
407. Similarly, CD8+ T-cells
from the earliest insulitic lesions are clearly cytotoxic to islet cells, and
show a recurrent amino acid sequence motif in the complementarily determining
region 3 of their α chains, with a prevalence of Vα17 frequently
joined to the Jα42 segment, although there is a diverse TCR Cα and
Cβ repertoire408. The latter observation suggested
that the majority of CD8+ T-cells participating in the initial phase
of autoimmunity recognize a limited number of MHC class I-peptide complexes
expressed by β-cells, although subsequent studies indicate that more than
one antigenic specificity is present409. Nonetheless, it remains possible
that expansion of the autoreactive repertoire had already occurred prior to
these analyses being conducted. It is also possible that multiple autoantigens
participate in the initiation of the autoimmune response, but that disease
progression requires the subsequent activation of an autoresponse to a
"primary" (major but not initial) antigen such as proinsulin. Such an
hypothesis would reconcile the apparently conflicting facts that the earliest
insulitic lesions contain few if any T-cells that recognize proinsulin 173,
407, yet proinsulin 1-/- NOD
mice are almost all protected from disease. It must be noted, however, that a
few proinsulin 1-/- NOD mice do develop T1D, indicating that
though important, it is not the only key auto antigen in these animals, a
conclusion that was also reached in a recent study in which preproinsulin 2 was
expressed in NOD APCs under control of a modified invariant chain promotor.
However, the possibility that residual T1D in preproinsulin 1-/- NOD
mice required autoreactivity to proinsulin 2, and that diabetes in mice
tolerized to preproinsulin 2 stemmed from reactivity to preproinsulin 1, has
gained some support from a recent study in which a modified preproinsulin 2
transgene with a substitution within the immunodominant B:9-23 epitope was
expressed in NOD mice lacking expression of both wild-type preproinsulin genes.
Animals lacking the B:9-23 epitope do not develop diabetes with suppressed
insulitis and insulin autoantibodies, highlighting the importance of this
epitope in disease induction in these animals70,
75. In addition, tolerance to
proinsulin blocks all NOD diabetes and expression of IGRP reactive T-cells.
The primacy of any one autoantigen in triggering human T1D is a matter
of debate. Serological studies of young at-risk children indicate that, on
average, autoimmunity to insulin appears before GAD65 which in turn precedes
IA-2 /phogrin autoantibodies, but only by a matter of months 296,
410. At the level of the individual,
however, any of these three autoantibodies may present first, and in older
subjects insulin autoantibodies appear only in a minority of patients at
disease onset 411,
412. Even if there is a primary
autoantigen, it is reasonable to hypothesize that any molecule that is a major
target of autoimmunity in type 1 diabetes is a candidate for use in
antigen-based therapy. Effective tolerization strategies in NOD mice based on
immunization with the native epitopes of insulin, or GAD65 or insulin B-chain
cDNA 413, appear to bear this out, although
whether these therapies can be effectively transferred to human subjects
remains a matter of debate 414.
Epitope spreading
and the progression to overt disease
Although invasive insulitis appears to be a necessary
precursor to disease, overt T1D does not necessarily occur even in the presence
of profound lymphocytic accumulation 415. Histologic studies indicate that
beta cell loss in the NOD mouse proceeds over a considerable time period,
namely beta cell destruction is chronic and likely balanced by beta cell
proliferation 416,
417. Experiments using congenic mouse
strains have suggested that passage through the final barrier that precedes
clinical disease (checkpoint 2) is mainly under control of non-MHC loci 57,
418, whilst those using transgenic
BDC2.5 TCR mice have demonstrated that epitope spreading is not a pre-requisite
(although it may be important in the “natural” disease419). Although expansion of the immune
response to include novel targets, and avidity maturation of individual
clonotypes 352 may play an important role, recent
studies have suggested that the transition to the destructive phase is
dependent upon the breakdown of key regulatory mechanisms, including both
decreased sensitivity of diabetogenic cells to negative signaling 420,
421, and functional depletion of
regulatory T-cell populations422,
423. Subsequently the breakdown in
peripheral tolerance might lead to further changes in the Th1/Th2 balance 424-427 (though fewer), up-regulation of
T-cell effector molecules (such as FasL 428 or inflammatory cytokines),
recruitment of additional effector cells (including activated macrophages and
cytotoxic T-cells 429), or cytokine-driven changes in
ß-cell function (such as up-regulation of MHC class I molecules 430 or increased sensitivity to
oxidative damage 431,
432) (Figure 2), all of which might
contribute to the uncontrolled destruction of pancreatic ß-cells. Though in the NOD mouse checkpoint 2 is often
discussed as a relatively acute event in both men and mouse, there is evidence
for asynchronous destruction of beta cells within individual islets over time
and in man, metabolic deterioration usually occurs over years 1,
433.
Multiple interventions can prevent the progression to T1D in
neonatal NOD mice, an increasing list of agents are effective once checkpoint 2
has been passed. Transplantation with sustained immunosuppression, non-specific
T-cell depleting or modulating antibodies and fusion proteins have proven
effective for long-term abrogation of established disease in mice (reviewed by434). Successful agents include
polyclonal anti-lymphocyte serum 26164}314, non-depleting anti-CD3 46,
47, sICAM/Ig 315, and depleting anti-CD4 with 435, or without 316, anti-CD8. In each case the studies
indicate a critical window of effectiveness, namely the first 7-14 days
following overt hyperglycemia, and cannot provide long-term protection for
allografts introduced into spontaneously diabetic animals. Reversal of new
onset diabetes has been achieved recently with multiple agents and experimental
designs emphasizing treatment with the initial increase in glucose immediately
after the onset of hyperglycemia. Therapies have included tyrosine kinase
inhibitors 436, injection of substance P 437, antithymocyte globulin, adaptive T
regulatory cells 438, exendin-4 and lysofylline439, dendritic cells 440, and insulin coupled to antigen
presenting cells 441.
The success of the anti-CD3 therapy in NOD mice has led to
recent clinical trials. The first, using a non-mitogenic derivative of OKT3
(OKT31 Ala Ala; 442preserved ß-cell mass in 9/12
newly-diabetic subjects in the 12 months following diagnosis 20,
443.The effect was maintained for at
least 2 years, although it diminished during the second year of follow-up 318. The second trial, using an
aglycosylated derivative of a rat antibody (ChAglyCD3;444) also preserved residual ß-cell
function for at least 18 months26167}, although in this case efficacy was
limited to a subgroup of the treated patients who had the highest initial
residual function. In both humans and mice anti-CD3 therapy likely functions in
2 phases, initially acting to clear the
insulitis, and subsequently decreasing the effector : regulatory T-cell ratio
to promote long-term tolerance 445,
446. Larger phase III trials have been
less encouraging21.
Interestingly, expansion of IL-10 producing proinsulin-specific T-cells
was observed in a case of spontaneous remission of T1D following insulin
therapy 447. A complete understanding of the
therapeutic mechanism of anti-CD3 treatment is still lacking, but the insight
so far gained provides the basis for improved strategies to increase
effectiveness in human subjects. For example, clearing of the insulitis
presumably prevents further autoimmune ß-cell damage, and, likely at least in
part due to the alleviation of cytokine-mediated inhibition of insulin
secretion, allows a rapid restoration of normoglycemia, reducing ß-cell stress,
and potentially allowing islet regeneration 322. Consequently, strategies to
combine T-cell depletion with agents to promote ß-cell growth may be more
effective than treatment with either agent alone 448. Similarly, improved methods of
generating human antigen-specific regulatory T-cells either in situ or ex vivo should lead to a more effective therapy 323,
324.
Mechanism
and mediators of ß-cell death
At present, the precise molecular mechanisms that lead to
ß-cell death in the destructive phase of T1D remain largely unresolved. It is
generally accepted that death primarily occurs by apoptosis (316-319), but the relative contributions of
direct effector cell:ß-cell contact, and of
secreted or shed soluble mediators, in the destructive process are
matters of some debate 449,
450, and may vary between mouse and
human T1D 451. Thus, experiments investigating
the involvement of proteins involved in contact-dependent killing such as
perforin, Fas, or FasL have produced inconclusive results. For example, NODlpr mice (which are deficient in Fas
expression) are protected from spontaneous diabetes 330,
331, consistent with a critical role
for Fas-FasL interactions in ß-cell killing. However, as NODlpr animals show little insulitis it is
possible that the lymphadenopathy seen in these animals significantly alters
their T-cell responses to diabetic autoantigens, or that Fas is required for
priming of the autoresponse, but is redundant at later times 452. Similarly, IL-1ß mediated
induction of Fas by ß-cells can be demonstrated in vitro 333,
334, but is not seen in spontaneous T1D
in NOD mice 453, although ß-cell expression of Fas
is observed in inflamed human islets 334,
454, and is readily detectable
following accelerated T1D in mice335,
336. Likewise, perforin-deficient NOD
mice show a reduced incidence and delayed onset of spontaneous disease 455, but perforin-deficient NY8.3 TCR
transgenic NOD mice develop diabetes more frequently than their
perforin-competent littermates 338.
Similarly, despite circumstantial evidence supporting a role
for NO-mediated oxidative damage456 in triggering ß-cell death (e.g.456, iNOS is not essential for diabetes
in NOD mice 457 or for cytokine-mediated
destruction of human islets458. Some of the apparent discrepancies
between individual studies may stem from the fact that most of the effectors
tested have pleiotrophic, and often conflicting, effects. For example, NO may
be directly cytotoxic to ß-cells, but also favors the generation of a Th2
biased immune response, which is likely to be less pathogenic to islets, whilst
ligation of Fas can trigger either apoptosis or proliferation of target cells
depending upon context 459. Alternatively the aforementioned
inconsistencies may simply reflect functional redundancy within the immune
system, and the likelihood that in the natural disease terminal ß-cell
destruction involves multiple effector mechanisms, which can compensate for
each other's absence. However, it is also possible that the key effector
mechanism for ß-cell death in T1D remains to be elucidated.
T-cell subsets in T1D
In
both humans and NOD mice multiple cell-types are recruited to islet infiltrates
during the progression to T1D, including CD4+ and CD8+
T-cells, B cells, macrophages, dendritic cells, and NK cells 460-462. In NOD mice macrophages and
dendritic cells precede T lymphocytes into the islet 463, and play essential roles in the
disease process. Presumably this is also true for the human disease 364. However, as stated above, T-cells
are considered to be the primary mediators of T1D in both humans and mice. The relative importance of the CD4+
and CD8+ subsets in the diabetogenic process has been a matter of
some debate 121,
464. Both CD4+ and CD8+
T-cells are required for adoptive transfer of disease from diabetic NOD mice to
irradiated hosts 49,
365, and antibodies to either subset
can prevent disease at early time points 366,
367, 373. However, whereas treatment of NOD
mice with antibodies to CD4 can arrest T1D progression at late stages 465, and may even reverse established
disease 316, anti-CD8 therapy is only effective
when given to young (2-5 week old) animals 466. Moreover, diabetes can be
transferred to NOD.scid mice lacking
expression of MHC class I by splenocytes from overtly diabetic, but not
pre-diabetic donors369,
370, and in several instances to
immuno-compromised animals by individual CD4+ T-cell clones (for
example50,
54). This suggests that CD8+
T-cells are essential for the initiation of diabetogenesis, but are dispensable
for the destructive phase 466. However, most NOD mice selectively
deficient in pancreatic ß-cell expression of MHC class I molecules, due to
co-transgenesis for human insulin promotor driven Cre and loxP flanked ß2-microglobulin,
develop insulitis but not hyperglycemia, challenging this conclusion.
Despite
their obvious importance to diabetes induction in un-manipulated NOD mice, CD4+
T-cells are dispensable for disease induction in some other animal models of
T1D. Thus, in contrast to several other CD8-restricted TCR transgenic mice,
those expressing the TCR of the AI4 CTL develop spontaneous disease in the
absence of CD4+ T-cells 372. Similarly, RIP-LCMV mice that do
not express the transgene in their thymuses rapidly develop T1D following LCMV
infection even when depleted of CD4+ T-cells. Together, the mouse
models suggest that circumstances can be contrived under which either subset is
unnecessary. However, it seems most reasonable to conclude that in the
“natural” disease both subsets contribute significantly to diabetogenesis, and
that treatment of established disease will likely require modulation of both
CD4+ and CD8+ restricted T-cell responses.
In mice, the CD4+ T-cell subset can be broadly classified into 6
subgroups on the basis of cytokine secretion profile and effector function,
namely Th1, Th17, Th2, Th3, Tr1, and Treg. Th1, which secrete
IFNγ, TNF-a and IL-2, and Th17 (IL17) are
primarily associated with cellular immunity, and Th1 has been strongly
implicated in diabetogenesis373-375. Moreover, ectopic islet expression
of the signature cytokines can promote the development of T1D (for example 98,
104, 376 but see also377), and the systemic treatment of NOD
mice with IL-12, a potent inducer of Th1 cells, leads to accelerated diabetes 378,
379. In contrast, Th2 cells, which
secrete IL-4, IL-5, IL-6, IL-9, IL-10 and IL-13, and are mainly involved in
humoral immune responses and allergy 467, seem either to be relatively
innocuous 426, or to provide protection from T1D 381-385,
385, 387, 388, and it is generally believed that
an increase in the Th1:Th2 ratio promotes diabetogenesis 424-427. Consistent with this hypothesis,
RIP-IL-4 NOD mice that over-express the signature cytokine in their islets are
protected from diabetes468. However, NOD mice deficient in
IL-4 (the primary polarizing cytokine for Th2 development) exhibit similar
disease incidences to regular NOD mice 469.
Transfer experiments with Th2 cells generated from BDC2.5 transgenic
mice have provided conflicting results. Naive T-cells cultured in the presence
of IL-4 and an anti-IFN-γ monoclonal antibody for 4-6 days and then
transferred to neonatal NOD recipients, invaded the islets, but neither
provoked disease, nor provided significant protection from co-injected Th1
polarized cells 426, although they did induce diabetes
when transferred to NOD.scid animals 470. However, in the latter case the
inflammatory lesion resembled an allergic inflammation, and the kinetics of
disease were markedly different from T1D induced by Th1 cells. In contrast,
naive BDC2.5 T-cells cultured for an extended period under Th2 polarizing
conditions, and subsequently cloned, generated effectors capable of
accelerating disease in neonatal NOD recipients, but which were unable to
induce diabetes in NOD.scid mice
unless a second Th1 polarized ß-cell specific T-cell clone was co-transferred 361. The reason for these obviously
contradictory results is not immediately apparent, but might reflect
differences in the relative amounts of the various Th2 cytokines generated by
the T-cells polarized under the 2 conditions. Thus, the diabetes induced in
NOD.scid mice by Th2 polarized
T-cells could be prevented by treatment with neutralizing antibodies to IL-10,
but not IL-4, suggesting that it might share some features with the accelerated
T1D observed in RIP-IL10 NOD mice, which also exhibit an atypical islet infiltrate
471. In contrast, T1D induced by the
BDC2.5 Th2 polarized clones may reflect IL-4 mediated
activation of APCs, and their consequent shift from a tolerizing to priming
state 392. Together, these results suggest
that under specific circumstances Th2 polarized T-cells are also capable of
promoting diabetogenesis, which raises important concerns regarding potential
therapies based upon altering the Th1/Th2 balance or administration of Th2
cytokines, particularly for treating lymphopenic individuals. Nonetheless,
numerous studies using NOD mice have indicated a beneficial effect of
increasing systemic IL-4 and/or IL-10 levels through administration of
recombinant protein or gene therapy (for example 381,
385, 393-399). At present this therapy has not
been tried in pre-diabetic patients, and it must be noted that the strict
dichotomy between IFNγ and IL-4 or IL-10 secretion by mouse CD4+
T-cell subsets is not recapitulated in the human immune system.
Both Th1 and Th2 subsets can properly be regarded as immune
effectors. However, it is now generally accepted that multiple populations of
regulatory T-cells also exist, and are critical to maintaining peripheral
tolerance (reviewed in472-474. Thus, the outcome of any immune
response, including autoimmunity, depends upon the balance between activated
effector and suppressor cells at critical anatomical sites 475. Since a numerical or functional
deficiency in regulatory T-cells will confer susceptibility to autoimmunity,
considerable recent interest has been focused on defining their properties and
ontogeny. Operationally, regulatory T-cells cells can be divided into two basic
classes; innate and induced 476. The repertoire and numbers of the
innate class, which include NKT cells (reviewed by 477 and Foxp3 expressing CD4+CD25hi Treg
cells (reviewed by 478, may be genetically determined 36,
153, 479, 480, although this remains a matter of
debate481,
482. Nonetheless, peripheral 476 deficiencies in both NKT and
resting CD4+CD25+ T-cells have been reported in human
diabetic subjects, and NOD mice are deficient in NKT cells402. The spontaneous population of Foxp3 expressing CD4+CD25hi
Treg cells develop in the thymus, and are presumably specific to
self-antigens expressed by the thymic epithelium or its resident APCs483. Thus, abnormally low thymic
expression of a peripheral antigen, such as may occur in individuals with class
I preproinsulin promoter VNTR alleles (the IDDM2 locus)225,
228, may confer disease susceptibility
either by decreased production of organ-specific Treg cells,
defective negative selection, or by a combination of these 2 probable
consequences. A modest numerical deficiency of CD4+CD25hi
Treg cells in NOD mice has been reported 403, although this conclusion was
subsequently disputed following comparisons with a wider range of
non-autoimmune prone strains 481. However, more recently an
age-dependent decline in the proportion of FoxP3+ CD4+CD25hi
Treg cells in the PLN and islet infiltrates of female NOD mice was
reported, suggesting a defect in their maintenance and/or expansion in these
animals 404.
Besides constitutive expression of CD25, several other
surface markers have been used to define spontaneous regulatory T-cells
including CD62L, CD45RBlo, and CD103339,
340. However, there is heterogeneity
for these markers within the CD4+CD25hi subset,
suggesting that multiple populations of Treg cells are generated,
and that these may differentially protect particular organs341. In this regard it should be noted
that protection of BDC2.5NOD.scid
mice from spontaneous T1D was provided by transfer of polyclonal CD4+CD62L+
T-cells rather than by polyclonal CD4+CD25+ T-cells 484, although in vitro expanded monoclonal BDC2.5 CD4+CD25hi
T-cells can also prevent diabetes in other systems 180,
181. Foxp3 expressing Treg cells can also be generated from
peripheral CD4+CD25- cells by activation in the presence
of TGF-ß 343,
344, although whether the precursors are
truly naive or are pre-programmed in the thymus, remains a matter of discussion
(reviewed by 485). In addition to Foxp3 expressing Treg cells,
at least 2 other subsets of inducible, antigen-specific, CD4+ regulatory
T-cells are found in the periphery, namely Th3 and Tr1 cells. The former subset
provides "help" for IgA production, mainly secretes TGF-ß, and is
primarily involved in establishing mucosal tolerance 486,
487. In contrast, Tr1 cells secrete
multiple cytokines including IL-10, IL-5, IFNa and TGF-ß, but little IL-2 or IL-4 488, and are induced during chronic
antigenic exposure. Tr1 cells can also be induced by some subsets of
"tolerogenic" APCs 489, and by Treg cells 490, and regulatory cells mediate the
long-term protective effects of anti-CD3 therapy 445 in NOD mice. Both Th3 and Tr1 cells secrete
immunosuppressive cytokines and can cause bystander suppression, although they
can also act by a contact-mediated mechanism. The innate CD4+CD25hi Treg cells, by contrast, exert
their suppressive effect in a cytokine-independent fashion, but can act both
directly upon activated T-cells, as well as indirectly through direct effects
on APCs 491,
492. In addition to regulatory CD4+
T-cells, subsets of CD8+ suppressor T-cells have also been
identified 492,
493, and may play a role in
CD3-mediated preservation of ß-cell mass in humans 347.
Although CD4+CD25hi Treg
cells were initially regarded as being anergic to in vitro stimulation, conditions have since been defined allowing
their ex vivo expansion (for example 180), and it is now clear that
they can proliferate extensively in vivo
348, 349, 403. Moreover, the fact that they are able to control an
ongoing autoimmune response suggests that the transfer of autologous ß-cell
specific Treg cells represents a rational strategy for therapeutic
intervention in pre-diabetic and possibly new-onset diabetic, subjects494. However, the low frequency of antigen-specific Treg
cells in peripheral blood, and the need to carefully select the target
antigen(s) to reduce the risk of unwanted side-effects such as increased tumor
susceptibility 495 provide important caveats. Similarly, although
treatment with the NKT cell ligand a-galactosyl
ceramide can protect NOD mice from T1D 352, 353, doubts have been expressed about its efficacy in
humans unless organ-specific NKT cells can be identified and targeted 496.
It is clear that B-lymphocytes and potentially
antibodies contribute to the development of diabetes of the NOD mouse and
likely man 322. Anti-CD20 therapy of NOD mice prevents diabetes
associated with induction of regulation and in new onset patients delays loss
of c-peptide 497.
Problems associated with the detection of T-cell reactivity to ß-cell
antigens in human subjects - Conclusions from International T-Cell Workshops
There
is a considerable amount of direct evidence that T-cells are primary mediators
of ß-cell destruction in the NOD mouse. In contrast, there is much less
certainty that this is also the case in human subjects, with much of the
available evidence being circumstantial. A major obstacle to characterizing
ongoing islet-specific T-cell responses in humans comes from the fact that the
only practical source of starting material is peripheral blood. However, it is
generally accepted that the frequency of islet-specific T-cells in the
peripheral blood of prediabetic or newly diabetic patients is very low 498, and newly diagnosed T1D patients
are often lymphopenic 499. Moreover, there is relatively high
percentage of monocytes, which may influence in vitro proliferation assays. Consequently it is technically
difficult to isolate and expand auto-antigenic T-cells, although the recent
appreciation that the effector and regulatory subsets have overlapping, or even
identical, specificities, suggests that the actual precursor
frequencies might be higher than originally appreciated 500. Despite the technical challenges,
many investigators have obtained evidence that diabetic subjects have T-cell
reactivity to ß-cell associated antigens, and recent studies have suggested
that examination of autoimmunity within different T-cell subsets might provide
insight into the T-cell responses of different clinical groups of patients.
Thus, in newly diagnosed T1D patients autoimmune T-cell responses were
primarily present amongst activated T-cells (CD45RA+RO+),
whereas those with longer disease duration reacted to autoantigens with memory
cells (CD45RO+).
The
first international workshop for the standardization of the T-cell assays
organized by the Immunology of Diabetes Society in 1999 aimed to identify, and
suggest solutions to, the problems associated with autoreactive T-cell assays
in T1D 501. The workshop defined a series of
candidate autoantigens that were distributed to 26 participating laboratories
around the world where T-cell proliferative responses of diabetic and
non-diabetic individuals were analyzed worldwide in 26 laboratories. Three
conclusions were reached:
1)
The quality of the recombinant antigens required improvement.
2)
All laboratories were able to detect T-cell responses to the control antigen
(tetanus toxoid) although with significant inter-laboratory variation in
sensitivity.
3)
Significant differences in T-cell proliferative responses to diabetic autoantigens
between diabetics and non-diabetic individuals were not consistently observed.
To
follow-up this initial study a second international T-cell workshop was held
which focused on the identification of more suitable antigens and the
development of standardized assays. Various preparations of GAD65,
proinsulin, and IA-2 were evaluated for endotoxin content, their ability to
stimulate T-cell clones, and any inhibitory effects they had on proliferation
to control antigens. Subsequently, recommendations were made for the
preparation of antigens with optimal quality.
T-cell assay development.
The
difficulties associated with human studies using T-cell assays based upon
proliferation have stimulated the development of alternative procedures, with
considerable effort being devoted to developing ELISPOT and tetramer based
assays. The former technique is capable of defining both the frequency and
cytokine profile of autoresponsive cells, and was the focus of the third
international T-cell workshop that demonstrated its potential for detecting
low-level autoreactive T-cell responses502. For example, ELISPOT analysis has
provided evidence for IFNγ-producing insulin B:9-23 reactive T-cells in
peripheral blood from new onset and prediabetic patients. These
observations were confirmed in the recent fourth international T-cell workshop
that used a panel of coded samples and antigens in a fully blinded study.
Synthetic
tetramers in which biotinylated MHC class I or class II molecules loaded with a
specific T-cell epitope are multimerized using fluorescent streptavidin provide
another tool to follow autoreactive lymphocytes. Thus, H-2Kd MHC
class I tetramers combined with insulin B:15-23 or NRP-V7, a mimetope peptide
to the NY8.3 CTL clone, have been used to study CD8+ T-cells in NOD
insulitic lesions. Interestingly, B:15-23 reactive T-cells were readily
detectable in islet infiltrates at an early age (4-5 weeks) but not later,
while NRP-V7 reactive T-cells were found only in older mice (11-18
weeks). Using the NY8.3 mimetope peptide evidence has been obtained
for “avidity maturation” of the CD8+ T-cell response to islets
during the development of disease. Moreover, analysis of NRP-V7-reactive
T-cells in peripheral blood from pre-diabetic NOD mice revealed a significant
increase immediately prior to disease onset, providing the first assay capable
of accurate temporal prognosis of T1D 350. In contrast, B:15-23 reactive CD8+
T-cells were not detected in peripheral blood.
CD4+
T-cells can also be analyzed by tetramers. When the specific target antigen of
the BDC2.5 T-cell clone was unknown, tetramers combining I-Ag7 with
mimetopes of the peptide for the BDC2.5 TCR efficiently stained both BDC2.5
transgenic T-cells, and thymic CD4+ T-cells from NOD mice 503-505. In contrast, T-cells recognizing
I-Ag7 tetramers loaded with GAD65 peptides could only be detected in
immunized mice 506, although HLA-DRB1*0401 tetramers
containing an immunodominant peptide from GAD65 can detect CD4+
autoreactive T-cells in peripheral blood of T1D patients 417,
419. With register trapping239 Kappler’s laboratory ahs developed
tetramers recognizing insulin B:9-23 T cells of the NOD mouse241. The possibility of using tetramers
for therapeutic, as well as diagnostic, applications is also being actively
pursued 507.
A very interesting approach is the use of nanoparticles with MHC-peptide
immunization generating CD8 T cells that can kill antigen presenting cells
expressing islet autoantigens213.
Summary
The
NOD mouse has been available to research community for over 20 years and in that
time tremendous advances have been made in understanding the role of the immune
system in development of T1D. It is clear that ß-cell destruction is due to an
adaptive immune response with both CD4+ and CD8+ T-cells
playing important roles. Moreover, NOD mice, as well as other rodent models of
T1D, have provided key insights into how tolerance can be broken and restored.
It should be kept in mind that development of diabetes in the NOD mouse differs
from that in humans in several respects, including a marked sex bias in females
and very high concordance (80-90% in NOD female mice). Nevertheless, the NOD
mouse provides a very important model for understanding diabetes pathogenesis
in humans, and for developing potential therapeutic strategies. There is a
developing consensus that the immune system response to insulin may be primary
with elimination of other target antigens dispensable for disease. If there is
a primary autoantigen and epitope (e.g. insulin B:9-23) understanding at a
structural level, the trimolecular complex (I-A87 – peptide – TCR)
may lead to novel deletional therapies. Even if there is a primary antigen
induction of T regs to multiple islet antigens, with suppression of
autoimmunity can clearly suppress disease. Thus, multiple different antigen
specific therapies are candidates for diabetes prevention.
The data obtained from investigations of human subjects, though not conclusive,
are highly suggestive of a direct role for T-cells in initiating ß-cell
destruction, although it remains uncertain to what extent data from the various
mouse models can be extrapolated to the human condition. However, it is
hopefully only a matter of time before advances in techniques will allow a
definitive assessment of the roles of the various T-cell subsets in human T1D,
and the development of effective strategies for accurate diagnosis and
antigen-specific therapeutic intervention to prevent or retard disease
progression.
Reference List
1. Eisenbarth
GS. Banting Lecture 2009: An Unfinished Journey: Molecular Pathogenesis to
Prevention of Type 1A Diabetes. diab 2010;59(4):759-774.
2. Gepts
W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. diab
1965;14(10):619-633.
3. Bottazzo
GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus
with autoimmune polyendocrine deficiencies. Lancet 1974;2(7892):1279-1283.
4. Nerup
J, Andersen OO, Bendixen G, Egeberg J, Poulsen JE. antipancreatic cellular
hypersensitivity in diabetes mellitus. diab 1971;20:424-427.
5. MacCuish
AC, Irvine WJ, Barnes EW, Duncan LJP. Antibodies to pancreatic islet cells in
insulin-dependent diabetics with coexistent autoimmune disease. Lancet
1974;2:1529-1531.
6. Gepts
W, LeCompte PM. The pancreatic islets in diabetes. Am J Med 1981;70(1):105-115.
7. Bottazzo
GF, Dean BM, McNally JM, Mackay EH, Swift PG, Gamble DR. In situ
characterization of autoimmune phenomena and expression of HLA molecules in the
pancreas in diabetic insulitis. N Engl J Med 1985;313(6):353-360.
8. Willcox
A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation
in human type 1 diabetes. Clin Exp Immunol 2009;155(2):173-181.
9. Velthuis
JH, Unger WW, Abreu JR et al. Simultaneous detection of circulating
autoreactive CD8+ T-cells specific for different islet cell-associated epitopes
using combinatorial MHC-multimers. diab 2010.
10. Sibley
RK, Sutherland DER, Goetz F, Michael AF. Recurrent diabetes mellitus in the
pancreas iso- and allograft. A light and electron microscope and
immunohistochemical analysis of four cases. Lab Invest 1985;53:132-144.
11. Sutherland
DE, Sibley R, Xu XZ et al. Twin-to-twin pancreas transplantation: reversal and
reenactment of the pathogenesis of type I diabetes. Trans Assoc Am Physicians
1984;97:80-87.
12. Lampeter
EF, Homberg M, Quabeck K et al. Transfer of insulin-dependent diabetes between
HLA-identical siblings by bone marrow transplantation. Lancet
1993;341:1243-1244.
13. Van
Vliet E, Roep BO, Meulenbroek L, Jan Bruining G, De Vries RRP. Human T cell
clones with specificity for insulinoma cell antigens. Eur J Immunol
1989;19:213-216.
14. Roep
BO, Arden SD, deVries RP, Hutton JC. T-cell clones from a type-1 diabetes
patient respond to insulin secretory granule proteins. Nature 1990;345:632-634.
15. Roep
BO, Kallan AA, Hazenbos WLW et al. T-cell reactivity to 38kD
insulin-secretory-granule protein in patients with recent-onset type 1
diabetes. Lancet 1991;337:1439-1441.
16. Neophytou
PI, Ozegbe P, Healey D, Quartey-Papafio R, Cooke A, Hutton JC. Development of a
procedure for the direct cloning of T-cell epitopes using bacterial expression
systems. J Immunol Methods 1996;196:63-72.
17. Arden
SD, Roep BO, Neophytou PI et al. Imogen 38: a novel 38-kD islet mitochondrial
autoantigen recognized by T cells from a newly diagnosed type I diabetic
patient. J Clin Invest 1996;97(2):551-561.
18. Kallan
AA, Roep BO, Arden SD, Hutton JC, de Vries RR. Beta-cell reactive T-cell clones
from type I diabetes patients are not beta cell specific and recognize multiple
antigens. J Autoimmun 1995;8(6):887-899.
19. Roep
BO. The role of T-cells in the pathogenesis of Type 1 diabetes: From cause to
cure. diabetol 2003;46(3):305-321.
20. Herold
KC, Hagopian W, Auger JA et al. Anti-CD3 monoclonal antibody in new-onset type
1 diabetes mellitus. N Engl J Med 2002;346(22):1692-1698.
21. Sherry
N, Hagopian W, Ludvigsson J et al. Teplizumab for treatment of type 1 diabetes
(Protege study): 1-year results from a randomised, placebo-controlled trial.
Lancet 2011;378(9790):487-497.
22. Bach
JF. Anti-CD3 antibodies for type 1 diabetes: beyond expectations. Lancet
2011;378(9790):459-460.
23. Like
AA, Butler L, Williams RM, Appel MC, Weringer EJ, Rossini AA. Spontaneous
autoimmune diabetes mellitus in the BB rat. diab 1982;31 (Suppl l pt 2):7-13.
24. Makino
S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a
non-obese, diabetic strain of mice. Exp Anim 1980;29:1-13.
25. Makino
S. Genetic analysis of IDDM in NOD mice. Exp Anim 1998;47(2):suppl-9.
26. Herold
KC, Bluestone JA. Type 1 diabetes immunotherapy: is the glass half empty or
half full? Sci Transl Med 2011;3(95):95fs1.
27. Driver
JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1
diabetes: a NOD to similarities and differences to human disease. Semin
Immunopathol 2011;33(1):67-87.
28. Roep
BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: re-evaluating the
use of animal models of type 1 diabetes. Nat Rev Immunol 2004;4(12):989-997.
29. Shoda
LK, Young DL, Ramanujan S et al. A comprehensive review of interventions in the
NOD mouse and implications for translation. Immunity 2005;23(2):115-126.
30. Serreze
DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of
antigen presenting cells in NOD/Lt mice. J Immunol 1993;150(6):2534-2543.
31. Rapoport
MJ, Lazarus AH, Jaramillo A, Speck E, Delovitch TL. Thymic T cell anergy in
autoimmune nonobese diabetic mice is mediated by deficient T cell receptor
regulation of the pathway of p21ras activation. J Exp Med
1993;177(4):1221-1226.
32. Serreze
DV, Leiter EH. Defective activation of T suppressor cell function in nonobese
diabetic mice. Potential relation to cytokine deficiencies. J Immunol
1988;140(11):3801-3807.
33. Carrasco-Marin
E, Shimizu J, Kanagawa O, Unanue ER. The class II MHC I-Ag7
molecules from non-obese diabetic mice are poor peptide binders. J Immunol
1996;156:450-458.
34. Kreuwel
HT, Biggs JA, Pilip IM, Pamer EG, Lo D, Sherman LA. Defective CD8+ T cell
peripheral tolerance in nonobese diabetic mice. J Immunol
2001;167(2):1112-1117.
35. Atkinson
MA, Bluestone JA, Eisenbarth GS et al. How does type 1 diabetes develop?: the
notion of homicide or beta-cell suicide revisited. diab 2011;60(5):1370-1379.
36. Kukreja
A, Cost G, Marker J et al. Multiple immuno-regulatory defects in type-1
diabetes. J Clin Invest 2002;109(1):131-140.
37. Spatz
M, Eibl N, Hink S et al. Impaired primary immune response in type-1 diabetes.
Functional impairment at the level of APCs and T-cells. Cell Immunol
2003;221(1):15-26.
38. Lang
J, Bellgrau D. A T-cell functional phenotype common among autoimmune-prone
rodent strains. Scand J Immunol 2002;55(6):546-559.
39. Mestas
J, Hughes CC. Of mice and not men: differences between mouse and human
immunology. J Immunol 2004;172(5):2731-2738.
40. Roep
BO, Atkinson M. Animal models have little to teach us about type 1 diabetes: 1.
In support of this proposal. diabetol 2004;47(10):1650-1656.
41. Leiter
EH, von HM. Animal models have little to teach us about type 1 diabetes: 2. In
opposition to this proposal. diabetol 2004;47(10):1657-1660.
42. Serreze
DV, Chen YG. Of mice and men: use of animal models to identify possible
interventions for the prevention of autoimmune type 1 diabetes in humans. Trends
Immunol 2005;26(11):603-607.
43. Kanazawa
Y, Komeda K, Sato S, Mori S, Akanuma K, Takaku F. Non-obese-diabetic mice:
immune mechanisms of pancreatic B-cell destruction. diabetol 1984;27:113-115.
44. Miyazaki
A, Hanafusa T, Yamada K et al. Predominance of T lymphocytes in pancreatic
islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal
study. Clin Exp Immunol 1985;60(3):622-630.
45. Itoh
N, Hanafusa T, Miyazaki A et al. Mononuclear cell infiltration and its relation
to the expression of major histocompatibility complex antigens and adhesion
molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent
diabetes mellitus patients. J Clin Invest 1993;92:2313-2322.
46. Chatenoud
L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission
of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA
1994;91(1):123-127.
47. Chatenoud
L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly
diabetic NOD mice. J Immunol 1997;158(6):2947-2954.
48. Wicker
LS, Miller BJ, Mullen Y. Transfer of autoimmune diabetes mellitus with
splenocytes from non-obese diabetic (NOD) mice. diab 1986;35(8):855-60.
49. Bendelac
A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes
from diabetic NOD mice to healthy neonates.
Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med
1987;166:823-32.
50. Nagata
M, Yoon JW. Studies on autoimmunity for T-cell-mediated beta-cell destruction.
Distinct difference in beta-cell destruction between CD4+ and CD8+ T-cell
clones derived from lymphocytes infiltrating the islets of NOD mice. diab
1992;41(8):998-1008.
51. Wong
FS, Visintin I, Wen L, Flavell RA, Janeway CA. CD8 T cell clones from young
nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice
in the absence of CD4 cells. J Exp Med 1996;183:67-76.
52. Delong
T, Baker RL, Reisdorph N et al. Islet Amyloid Polypeptide Is a Target Antigen
for Diabetogenic CD4+ T Cells. diab 2011;60(9):2325-2330.
53. Stadinski
BD, Delong T, Reisdorph N et al. Chromogranin A is an autoantigen in type 1
diabetes. Nat Immunol 2010;11(3):225-231.
54. Haskins
K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T cell
clones from nonobese diabetic mice. Proc Natl Acad Sci USA 1989;86:8000-8004.
55. Bergman
B, Haskins K. Islet-specific T-cell clones of the NOD mouse respond to
B-granule antigen. diab 1994;43(2):197-203.
56. Katz
JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell
from genesis through pathogenesis. Cell 1993;74:1089-1100.
57. Gonzalez
A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of
diabetes progression. Immunity 1997;7(6):873-883.
58. Andre-Schmutz
I, Hindelang C, Benoist C, Mathis D. Cellular and molecular changes
accompanying the progression from insulitis to diabetes. Eur J Immunol
1999;29(1):245-255.
59. Kurrer
MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated
autoimmune diabetes. Proc Natl Acad Sci U S A 1997;94(1):213-218.
60. Luhder
F, Katz J, Benoist C, Mathis D. Major histocompatibility complex class II
molecules can protect from diabetes by positively selecting T cells with
additional specificities. J Exp Med 1998;187(3):379-387.
61. Burton
AR, Baquet Z, Eisenbarth GS et al. Central Nervous System Destruction Mediated
by Glutamic Acid Decarboxylase-Specific CD4+ T Cells. J Immunol 2010.
62. Serreze
DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major
histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and
insulitis resistant. diab 1994;43(3):505-509.
63. Katz
J, Benoist C, Mathis D. Major histocompatibility complex class I molecules are
required for the development of insulitis in non-obese diabetic mice. Eur J
Immunol 1993;23(12):3358-3360.
64. Sumida
T, Furukawa M, Sakamoto A et al. Prevention of insulitis and diabetes in beta
2-microglobulin-deficient non-obese diabetic mice. Int Immunol
1994;6(9):1445-1449.
65. Wicker
LS, Leiter EH, Todd JA et al. Beta 2-microglobulin-deficient NOD mice do not
develop insulitis or diabetes. diab 1994;43(3):500-504.
66. Hamilton-Williams
EE, Serreze DV, Charlton B et al. Transgenic rescue implicates
beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic
(NOD) mice. Proc Natl Acad Sci U S A 2001;98(20):11533-11538.
67. Perarnau
B, Siegrist CA, Gillet A, Vincent C, Kimura S, Lemonnier FA. Beta
2-microglobulin restriction of antigen presentation. Nature
1990;346(6286):751-754.
68. Moriyama
H, Abiru N, Paronen J et al. Evidence for a primary islet autoantigen
(preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse.
Proc Natl Acad Sci U S A 2003;100(18):10376-10381.
69. Thebault-Baumont
K, Dubois-LaForgue D, Krief P et al. Acceleration of type 1 diabetes mellitus
in proinsulin 2-deficient NOD mice. J Clin Invest 2003;111(6):851-857.
70. Nakayama
M, Abiru N, Moriyama H et al. Prime role for an insulin epitope in the
development of type 1 diabetes in NOD mice. Nature 2005;435(7039):220-223.
71. Oeser
JK, Parekh VV, Wang Y et al. Deletion of the G6pc2 Gene Encoding the
Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein Does Not
Affect the Progression or Incidence of Type 1 Diabetes in NOD/ShiLtJ Mice. diab
2011.
72. Kubosaki
A, Miura J, Notkins AL. IA-2 is not required for the development of diabetes in
NOD mice. diabetol 2004;47(1):149-150.
73. Kubosaki
A, Gross S, Miura J et al. Targeted disruption of the IA-2beta gene causes
glucose intolerance and impairs insulin secretion but does not prevent the
development of diabetes in NOD mice. diab 2004;53(7):1684-1691.
74. Kash
SF, Condie BG, Baekkeskov S. Glutamate decarboxylase and GABA in pancreatic
islets: lessons from knock-out mice. Horm Metab Res 1999;31(5):340-344.
75. Nakayama
M, Beilke JN, Jasinski JM et al. Priming and effector dependence on insulin
B:9-23 peptide in NOD islet autoimmunity. J Clin Invest 2007;117(7):1835-1843.
76. Krishnamurthy
B, Mariana L, Gellert SA et al. Autoimmunity to Both Proinsulin and IGRP Is
Required for Diabetes in Nonobese Diabetic 8.3 TCR Transgenic Mice. J Immunol
2008;180(7):4458-4464.
77. Krishnamurthy
B, Dudek NL, McKenzie MD et al. Responses against islet antigens in NOD mice
are prevented by tolerance to proinsulin but not IGRP. J Clin Invest
2006;116(12):3258-3265.
78. Holst
J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA.
Generation of T-cell receptor retrogenic mice. Nat Protoc 2006;1(1):406-417.
79. Burton
AR, Vincent E, Arnold PY et al. On the pathogenicity of autoantigen-specific
T-cell receptors. Diabetes 2008;57(5):1321-1330.
80. Adams
TE, Apert S, Hanahan D. Non-tolerance and autoantibodies to a transgenic
self-antigen expressed in pancreatic beta cells. Nature 1987;325:223-228.
81. Jolicoeur
C, Hanahan D, Smith KM. T-cell tolerance toward a transgenic b-cell antigen and transcription of endogenous
pancreatic genes in thymus. Proc Natl Acad Sci USA 1994;91(14):6707-6711.
82. Mathis
D, Benoist C. Back to central tolerance. Immunity 2004;20(5):509-516.
83. Forster
I, Hirose R, Arbeit JM, Clausen BE, Hanahan D. Limited capacity for
tolerization of CD4+ T cells specific for a pancreatic beta cell neo-antigen.
Immunity 1995;2(6):573-585.
84. Heath
WR, Allison J, Hoffmann MW et al. Autoimmune diabetes as a consequence of
locally produced interleukin-2. Nature 1992;359(6395):547-549.
85. Heath
WR, Karamalis F, Donoghue J, Miller JF. Autoimmunity caused by ignorant CD8+ T
cells is transient and depends on avidity. J Immunol 1995;155(5):2339-2349.
86. Kurts
C, Sutherland RM, Davey G et al. CD8 T cell ignorance or tolerance to islet
antigens depends on antigen dose. Proc Natl Acad Sci U S A
1999;96(22):12703-12707.
87. Kurts
C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility
complex class I-restricted cross-presentation is biased towards high dose
antigens and those released during cellular destruction. J Exp Med
1998;188(2):409-414.
88. Behrens
GM, Li M, Davey GM et al. Helper requirements for generation of effector CTL to
islet beta cell antigens. J Immunol 2004;172(9):5420-5426.
89. Kurts
C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class
I-restricted exogenous presentation of self antigens in vivo. J Exp Med 1996;184(3):923-930.
90. Li
M, Davey GM, Sutherland RM et al. Cell-associated ovalbumin is cross-presented
much more efficiently than soluble ovalbumin in vivo. J Immunol
2001;166(10):6099-6103.
91. Nikolic-Zugic
J, Bevan MJ. Role of self-peptides in positively selecting the T-cell
repertoire. Nature 1990;344(6261):65-67.
92. Kurts
C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted
cross-presentation of exogenous self-antigens leads to deletion of autoreactive
CD8(+) T cells. J Exp Med 1997;186(2):239-245.
93. Kurts
C, Heath WR, Kosaka H, Miller JF, Carbone FR. The peripheral deletion of
autoreactive CD8+ T cells induced by cross-presentation of self-antigens
involves signaling through CD95 (Fas, Apo-1). J Exp Med 1998;188(2):415-420.
94. Kurts
C, Carbone FR, Barnden M et al. CD4+ T cell help impairs CD8+ T cell deletion
induced by cross-presentation of self-antigens and favors autoimmunity. J Exp
Med 1997;186(12):2057-2062.
95. Schuurhuis
DH, Ioan-Facsinay A, Nagelkerken B et al. Antigen-antibody immune complexes
empower dendritic cells to efficiently prime specific CD8+ CTL responses in
vivo. J Immunol 2002;168(5):2240-2246.
96. Oldstone
MB, Nerenberg M, Southern P, Price J, Lweicki H. Virus infection triggers
insulin-dependent diabetes mellitus in a transgenic model: role of anti-self
(virus) immune response. Cell 1991;65:319-331.
97. Ohashi
PS, Oehen S, Buerki K et al. Ablation of "tolerance" and induction of
diabetes by virus infection in viral antigen transgenic mice. Cell 1991;65:305-317.
98. Lee
MS, von HM, Reiser H, Oldstone MB, Sarvetnick N. Sensitization to self (virus)
antigen by in situ expression of murine interferon-gamma. J Clin Invest
1995;95(2):486-492.
99. von
Herrath MG, Guerder S, Lewicki H, Flavell RA, Oldstone MB. Coexpression of B7-1
and viral ("self") transgenes in pancreatic beta cells can break
peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity
1995;3(6):727-738.
100. von
Herrath MG, Dockter J, Oldstone MBA. How virus induces a rapid or slow onset
insulin-dependent diabetes mellitus in a transgenic model. Immunity
1994;1:231-242.
101. Slifka
MK, Blattman JN, Sourdive DJ et al. Preferential escape of subdominant CD8+ T
cells during negative selection results in an altered antiviral T cell
hierarchy. J Immunol 2003;170(3):1231-1239.
102. Sevilla
N, Homann D, von Herrath M et al. Virus-induced diabetes in a transgenic model:
role of cross-reacting viruses and quantitation of effector T cells needed to
cause disease. J Virol 2000;74(7):3284-3292.
103. Lang
KS, Recher M, Junt T et al. Toll-like receptor engagement converts T-cell
autoreactivity into overt autoimmune disease. Nature Medicine
2005;11(2):138-145.
104. von
Herrath MG, Allison J, Miller JFAP, Oldstone MBA. Focal expression of
interleukin-2 does not break unresponsiveness to "self" (viral)
antigen expressed in b cells but
enhances development of autoimmune disease (diabetes) after initiation of an
anti-self immune response. J Clin Invest 1995;95:477-485.
105. Rhode
A, Pauza ME, Barral AM et al. Islet-specific expression of CXCL10 causes
spontaneous islet infiltration and accelerates diabetes development. J Immunol
2005;175(6):3516-3524.
106. von
HM, Holz A. Pathological changes in the islet milieu precede infiltration of
islets and destruction of beta-cells by autoreactive lymphocytes in a
transgenic model of virus-induced IDDM. J Autoimmun 1997;10(3):231-238.
107. Christen
U, von Herrath MG. Induction, acceleration or prevention of autoimmunity by
molecular mimicry. Mol Immunol 2004;40(14-15):1113-1120.
108. Filippi
C, von Herrath M. How viral infections affect the autoimmune process leading to
type 1 diabetes. Cell Immunol 2005;233(2):125-132.
109. Lo
D, Freedman J, Hesse S, Palmiter RD, Brinster RL, Sherman LA. Peripheral
tolerance to an islet cell-specific hemagglutinin transgene affects both CD4+
and CD8+ T cells. Eur J Immunol 1992;22(4):1013-1022.
110. Morgan
DJ, Kreuwel HT, Sherman LA. Antigen concentration and precursor frequency
determine the rate of CD8+ T cell tolerance to peripherally expressed antigens.
J Immunol 1999;163(2):723-727.
111. Piaggio
E, Hartemann-Heurtier A, Cabarrocas J et al. Maintaining or breaking CD8+
T-cell tolerance to beta islet cell antigens: lessons from transgenic mouse
models. J Autoimmun 2004;22(2):115-120.
112. Morgan
DJ, Kurts C, Kreuwel HT, Holst KL, Heath WR, Sherman LA. Ontogeny of T cell
tolerance to peripherally expressed antigens. Proc Natl Acad Sci USA
1999;96(7):3854-3858.
113. Mintern
JD, Sutherland RM, Lew AM, Shortman K, Carbone FR, Heath WR. Constitutive, but
not inflammatory, cross-presentation is disabled in the pancreas of young mice.
Eur J Immunol 2002;32(4):1044-1051.
114. Morgan
DJ, Liblau R, Scott B et al. CD8(+) T cell-mediated spontaneous diabetes in
neonatal mice. J Immunol 1996;157:978-983.
115. Degermann
S, Reilly C, Scott B, Ogata L, von Boehmer H, Lo D. On the various
manifestations of spontaneous autoimmune diabetes in rodent models. Eur J
Immunol 1994;24(12):3155-3160.
116. Scott
B, Liblau R, Degermann S et al. A role for non-MHC genetic polymorphism in
susceptibility to spontaneous autoimmunity. Immunity 1994;1(1):73-83.
117. Roman
LM, Simons LF, Hammer RE, Sambrook JF, Gething M-JH. The expression of
influenze virus hemagglutinin in the pancreatic b cells of
transgenic mice results in autoimmune diabetes. Cell 1990;61:383-396.
118. Abiru
N, Maniatis AK, Yu L et al. Peptide and major histocompatibility
complex-specific breaking of humoral tolerance to native insulin with the B9-23
peptide in diabetes-prone and normal mice. diab 2001;50(6):1274-1281.
119. Moriyama
H, Wen L, Abiru N et al. Induction and acceleration of insulitis/diabetes in
mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin
self-peptide. Proc Natl Acad Sci U S A 2002;99(8):5539-5544.
120. Havari
E, Lennon-Dumenil AM, Klein L et al. Expression of the B7.1 costimulatory
molecule on pancreatic beta cells abrogates the requirement for CD4 T cells in
the development of type 1 diabetes. J Immunol 2004;173(2):787-796.
121. Wong
FS, Karttunen J, Dumont C et al. Identification of an MHC class I-restricted
autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat
Med 1999;5(9):1026-1031.
122. Stewart
TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, Maclachlan NJ. Induction of
type I diabetes by interferon-alpha in transgenic mice. Science
1993;260(5116):1942-1946.
123. Banchereau
J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell
activation. Immunity 2004;20(5):539-550.
124. Tough
DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses
and type I interferon in vivo. Science 1996;272(5270):1947-1950.
125. Vezys
V, Lefrancois L. Cutting edge: inflammatory signals drive organ-specific
autoimmunity to normally cross-tolerizing endogenous antigen. J Immunol
2002;169(12):6677-6680.
126. Wen
L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes
and the expression of toll-like receptors on pancreatic islets. J Immunol
2004;172(5):3173-3180.
127. Horwitz
MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by
Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat
Med 1998;4(7):781-785.
128. Wong
S, Guerder S, Visintin I et al. Expression of the co-stimulator molecule B7-1
in pancreatic b-cells accelerates diabetes in the
NOD mouse. diab 1995;44:326-329.
129. Guerder
S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers
antigen-presenting-cell function to parenchymal tissue and in conjunction with
tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl
Acad Sci U S A 1994;91(11):5138-5142.
130. Karges
W, Pechhold K, Al Dahouk S et al. Induction of Autoimmune Diabetes Through
Insulin (but Not GAD65) DNA Vaccination in Nonobese Diabetic and in RIP-B7.1
Mice. diab 2002;51(11):3237-3244.
131. Andre
I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the
progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad
Sci U S A 1996;93(6):2260-2263.
132. Rainbow
DB, Esposito L, Howlett SK et al. Commonality in the genetic control of Type 1
diabetes in humans and NOD mice: variants of genes in the IL-2 pathway are
associated with autoimmune diabetes in both species. Biochem Soc Trans
2008;36(3):312-315.
133. Robles
DT, Eisenbarth GS, Dailey NJM, Peterson LB, Wicker LS. Insulin autoantibodies
are associated with islet inflammation but not always related to diabetes
progression in NOD congenic mice. diab 2002;52(3):882-886.
134. Maynard
J, Petersson K, Wilson DH et al. Structure of an autoimmune T cell receptor
complexed with class II peptide-MHC: insights into MHC bias and antigen
specificity. Immunity 2005;22(1):81-92.
135. Li
Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human
autoimmune TCR bound to a myelin basic protein self-peptide and a multiple
sclerosis-associated MHC class II molecule. EMBO J 2005;24(17):2968-2979.
136. Hahn
M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self
peptide-major histocompatibility complex binding by a human autoimmune T cell
receptor. Nat Immunol 2005;6(5):490-496.
137. Stadinski
B, Kappler J, Eisenbarth GS. Molecular targeting of islet autoantigens.
Immunity 2010;32(4):446-456.
138. Nicholson
MJ, Hahn M, Wucherpfennig KW. Unusual features of self-peptide/MHC binding by
autoimmune T cell receptors. Immunity 2005;23(4):351-360.
139. Mallone
R, Kochik SA, Reijonen H et al. Functional avidity directs T-cell fate in
autoreactive CD4(+) T cells. Blood 2005;106(8):2798-2805.
140. Liston
A, Lesage S, Gray DH et al. Generalized resistance to thymic deletion in the
NOD mouse; a polygenic trait characterized by defective induction of Bim.
Immunity 2004;21(6):817-830.
141. Skowera
A, Ellis RJ, Varela-Calvino R et al. CTLs are targeted to kill beta cells in
patients with type 1 diabetes through recognition of a glucose-regulated
preproinsulin epitope. J Clin Invest 2008;118(10):3390-3402.
142. Arif
S, Tree TI, Astill TP et al. Autoreactive T cell responses show proinflammatory
polarization in diabetes but a regulatory phenotype in health. J Clin Invest
2004;113(3):451-463.
143. Linn
T, Strate C, Federlin K, Papaccio G. Intercellular adhesion molecule-1 (ICAM-1)
expression in the islets of the non-obese diabetic and low-dose
streptozocin-treated mouse. Histochemistry 1994;102(4):317-321.
144. Hanninen
A, Taylor C, Streeter PR et al. Vascular addressins are induced on islet
vessels during insulitis in nonobese diabetic mice and are involved in lymphoid
cell binding to islet endothelium. J Clin Invest 1993;92(5):2509-2515.
145. Faveeuw
C, Gagnerault MC, Lepault F. Expression of homing and adhesion molecules in
infiltrated islets of Langerhans and salivary glands of nonobese diabetic mice.
J Immunol 1994;152(12):5969-5978.
146. Baron
JL, Reich EP, Visintin I, Janeway CA, Jr. The pathogenesis of adoptive murine
autoimmune diabetes requires an interaction between alpha 4-integrins and
vascular cell adhesion molecule-1. J Clin Invest 1994;93(4):1700-1708.
147. Hasegawa
Y, Yokono K, Taki T et al. Prevention of autoimmune insulin-dependent diabetes
in non-obese diabetic mice by anti-LFA-1 and anti-ICAM-1 mAb. Int Immunol
1994;6(6):831-838.
148. Yang
XD, Karin N, Tisch R, Steinman L, McDevitt HO. Inhibition of insulitis and
prevention of diabetes in nonobese diabetic mice by blocking L-selectin and
very late antigen 4 adhesion receptors. Proc Natl Acad Sci U S A
1993;90(22):10494-10498.
149. Moser
B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol
2001;2(2):123-128.
150. Greening
JE, Tree TI, Kotowicz KT et al. Processing and presentation of the islet
autoantigen GAD by vascular endothelial cells promotes transmigration of
autoreactive T-cells. diab 2003;52(3):717-725.
151. Savinov
AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by
endothelial cells and chemoattraction are required for homing of
insulin-specific CD8+ T cells. J Exp Med 2003;197(5):643-656.
152. Turley
SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut
non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci U S A
2005;102(49):17729-17733.
153. Rosmalen
JG, Leenen PJ, Pelegri C, Drexhage HA, Homo-Delarche F. Islet abnormalities in
the pathogenesis of autoimmune diabetes. Trends Endocrinol Metab
2002;13(5):209-214.
154. Sheridan
JP, Marsters SA, Pitti RM et al. Control of TRAIL-induced apoptosis by a family
of signaling and decoy receptors. Science 1997;277(5327):818-821.
155. Horwitz
MS, Ilic A, Fine C, Rodriguez E, Sarvetnick N. Presented antigen from damaged
pancreatic beta cells activates autoreactive T cells in virus-mediated
autoimmune diabetes. J Clin Invest 2002;109(1):79-87.
156. Trudeau
JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell
apoptosis: a trigger for autoimmune diabetes? diab 2000;49(1):1-7.
157. Hugues
S, Mougneau E, Ferlin W et al. Tolerance to islet antigens and prevention from
diabetes induced by limited apoptosis of pancreatic beta cells. Immunity
2002;16(2):169-181.
158. Herold
KC. Achieving antigen-specific immune regulation. J Clin Invest
2004;113(3):346-349.
159. Hyoty
H, Taylor KW. The role of viruses in human diabetes. diabetol
2002;45(10):1353-1361.
160. Delovitch
TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes:
immune dysregulation gets the NOD. Immunity 1997;7(6):727-738.
161. O'Brien
BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by
macrophages from NOD mice is reduced. diab 2002;51(8):2481-2488.
162. Norris
JM, Barriga K, Klingensmith G et al. Timing of cereal exposure in infancy and
risk of islet autoimmunity. The Diabetes
Autoimmunity Study in the Young (DAISY). JAMA 2003;290(13):1713-1720.
163. Winer
S, Tsui H, Lau A et al. Autoimmune islet destruction in spontaneous type 1
diabetes is not beta-cell exclusive. Nat Med 2003;9(2):198-205.
164. Gagnerault
MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for
priming of beta cell reactive T cells in NOD mice. J Exp Med
2002;196(3):369-377.
165. Jaakkola
I, Jalkanen S, Hanninen A. Diabetogenic T cells are primed both in pancreatic
and gut-associated lymph nodes in NOD mice. Eur J Immunol
2003;33(12):3255-3264.
166. Chen
Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ T reg cells impinge
on autoimmune diabetes. J Exp Med 2005;202(10):1387-1397.
167. Hanninen
A, Jaakkola I, Jalkanen S. Mucosal addressin is required for the development of
diabetes in nonobese diabetic mice. J Immunol 1998;160(12):6018-6025.
168. Hoglund
P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of
autoimmune diabetes by developmentally regulated presentation of islet cell
antigens in the pancreatic lymph nodes. J Exp Med 1999;189(2):331-339.
169. Haskins
K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for
pancreatic islet antigen. Diabetes 1988;37(10):1444-1448.
170. Kassem
SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and
apoptosis in the developing normal human pancreas and in hyperinsulinism of
infancy. diab 2000;49(8):1325-1333.
171. Ziegler
AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of
developing type 1 diabetes-associated autoantibodies. JAMA
2003;290(13):1721-1728.
172. Larger
E, Becourt C, Bach JF, Boitard C. Pancreatic islet beta cells drive T
cell-immune responses in the nonobese diabetic mouse model. J Exp Med
1995;181(5):1635-1642.
173. Baker
FJ, Lee M, Chien YH, Davis MM. Restricted islet-cell reactive T cell repertoire
of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci U S A
2002;99(14):9374-9379.
174. Yang
Y, Charlton B, Shimada A, Dal Canto R, Fathman CG. Monoclonal T cells
identified in early NOD islet infiltrates. Immunity 1996;4(2):189-194.
175. Notkins
AL, Lernmark A. Autoimmune type 1 diabetes: resolved and unresolved issues. J
Clin Invest 2001;108(9):1247-1252.
176. Tian
J, Gregori S, Adorini L, Kaufman DL. The frequency of high avidity T cells
determines the hierarchy of determinant spreading. J Immunol
2001;166(12):7144-7150.
177. Amrani
A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of
autoimmune diabetes driven by avidity maturation of a T-cell population. Nature
2000;406(6797):739-742.
178. Peterson
LD, van der Keur M, de Vries RR, Roep BO. Autoreactive and immunoregulatory
T-cell subsets in insulin-dependent diabetes mellitus. diabetol
1999;42(4):443-449.
179. Homann
D, Holz A, Bot A et al. Autoreactive CD4+ T cells protect from autoimmune
diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity
1999;11(4):463-472.
180. Tang
Q, Henriksen KJ, Bi M et al. In vitro-expanded antigen-specific regulatory T
cells suppress autoimmune diabetes. J Exp Med 2004;199(11):1455-1465.
181. Tarbell
KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25(+) CD4(+) T cells, expanded
with dendritic cells presenting a single autoantigenic peptide, suppress
autoimmune diabetes. Journal of Experimental Medicine 2004;199(11):1467-1477.
182. Eisenbarth
GS, Moriyama H, Robles DT et al. Insulin Autoimmunity:
prediction/precipitation/prevention type 1A diabetes. Autoimmunity Reviews
2002;1:139-145.
183. Serreze
DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are
critical antigen-presenting cells for the initiation of T cell-mediated
autoimmune diabetes in nonobese diabetic mice. J Immunol 1998;161(8):3912-3918.
184. Martin
S, Wolf-Eichbaum D, Duinkerken G et al. Development of type 1 diabetes despite
severe hereditary B-lymphocyte deficiency. N Engl J Med 2001;345(14):1036-1040.
185. Wenzlau
JM, Liu Y, Yu L et al. A common nonsynonymous single nucleotide polymorphism in
the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes.
diab 2008;57(10):2693-2697.
186. Dang
M, Rockell J, Wagner R et al. Human type 1 diabetes is associated with T cell
autoimmunity to zinc transporter 8. J Immunol 2011;186(10):6056-6063.
187. Yu
L, Robles DT, Abiru N et al. Early expression of antiinsulin autoantibodies of
humans and the NOD mouse: evidence for early determination of subsequent
diabetes. Proc Natl Acad Sci USA 2000;97(4):1701-1706.
188. Bowie
L, Tite J, Cooke A. Generation and maintenance of autoantigen-specific CD8(+) T
cell clones isolated from NOD mice. J Immunol Methods 1999;228(1-2):87-95.
189. Nagata
M, Santamaria P, Kawamura T, Utsugi T, Yoon J-W. Evidence for the role of CD8+
cytotoxic T cells in the destruction of pancreatic b-cells in nonobese diabetic mice. J Immunol
1994;152:2042-2050.
190. Wegmann
DR, Shehadeh N, Lafferty KJ, Norbury-Glaser N, Gill RG, Daniel D. Establishment
of islet-specific T cell lines and clones from islet isografts placed in
spontaneously diabetic NOD mice. J Autoimmun 1993;6:517-527.
191. Tree
TI, O'Byrne D, Tremble JM et al. Evidence for recognition of novel islet T cell
antigens by granule-specific T cell lines from new onset type 1 diabetic
patients. Clin Exp Immunol 2000;121(1):100-105.
192. Malarkannan
S, Horng T, Shih PP, Schwab S, Shastri N. Presentation of out-of-frame
peptide/MHC class I complexes by a novel translation initiation mechanism.
Immunity 1999;10(6):681-690.
193. Engelhard
VH. Creating new peptide antigens by slicing and splicing proteins. Nat Immunol
2004;5(2):128-129.
194. Lieberman
SM, Evans AM, Han B et al. Identification of the {beta} cell antigen targeted
by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes.
Proc Natl Acad Sci U S A 2003;100(14):8384-8388.
195. Babad
J, Geliebter A, DiLorenzo TP. T-cell autoantigens in the non-obese diabetic
mouse model of autoimmune diabetes. Immunology 2010;131(4):459-465.
196. Wegmann
DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant
component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol
1994;24(8):1853-1857.
197. Daniel
D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production
profile and diabetogenic activity of insulin-specific T cell clones isolated
from NOD mice. Eur J Immunol 1995;25(4):1056-1062.
198. Halbout
P, Briand JP, Becourt C, Muller S, Boitard C. T cell response to preproinsulin
I and II in the nonobese diabetic mouse. J Immunol 2002;169(5):2436-2443.
199. Chen
W, Bergerot I, Elliott JF et al. Evidence that a peptide spanning the B-C
junction of proinsulin is an early autoantigen epitope in the pathogenesis of
type 1 diabetes. J Immunol 2001;167(9):4926-4935.
200. Congia
M, Patel S, Cope AP, De Virgiliis S, Sonderstrup G. T cell epitopes of insulin
defined in HLA-DR4 transgenic mice are derived from preproinsulin and
proinsulin. Proc Natl Acad Sci USA 1998;95(7):3833-3838.
201. Semana
G, Gausling R, Jackson RA, Hafler DA. T cell autoreactivity to proinsulin
epitopes in diabetic patients and healthy subjects. J Autoimmun
1999;12(4):259-267.
202. Alleva
DG, Crowe PD, Jin L et al. A disease-associated cellular immune response in
type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest
2001;107(2):173-180.
203. Durinovic-Bello
I, I, Boehm BO, Ziegler AG. Predominantly Recognized ProInsulin T Helper Cell
Epitopes in Individuals With and Without Islet Cell Autoimmunity. J Autoimmun
2002;18(1):55-66.
204. Mannering
SI, Morris JS, Stone NL, Jensen KP, Van Endert PM, Harrison LC. CD4+ T cell
proliferation in response to GAD and proinsulin in healthy, pre-diabetic, and
diabetic donors. Ann N Y Acad Sci 2004;1037:16-21.:16-21.
205. Kent
SC, Chen Y, Bregoli L et al. Expanded T cells from pancreatic lymph nodes of
type 1 diabetic subjects recognize an insulin epitope. Nature
2005;435(7039):224-228.
206. Mannering
SI, Harrison LC, Williamson NA et al. The insulin A-chain epitope recognized by
human T cells is posttranslationally modified. J Exp Med 2005;202(9):1191-1197.
207. Toma
A, Haddouk S, Briand JP et al. Recognition of a subregion of human proinsulin
by class I-restricted T cells in type 1 diabetic patients. Proc Natl Acad Sci U
S A 2005;102(30):10581-10586.
208. Hutchings
P, Cooke A. Protection from insulin-dependent diabetes mellitus affected by insulin
antigens in incomplete Freund's adjuvant depends on route of administration. J
Autoimmun 1998;11:127-130.
209. Harrison
LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces
regulatory CD8 gamma delta T cells that prevent murine insulin-dependent
diabetes. J Exp Med 1996;184(6):2167-2174.
210. Daniel
D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal
or subcutaneous administration of insulin peptide B-(9-23). Proc Natl Acad Sci
USA 1996;93(2):956-960.
211. Martinez
NR, Augstein P, Moustakas AK et al. Disabling an integral CTL epitope allows
suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin
Invest 2003;111(9):1365-1371.
212. Zekzer
D, Wong FS, Wen L et al. Inhibition of diabetes by an insulin-reactive CD4
T-cell clone in the nonobese diabetic mouse. diab 1997;46(7):1124-1132.
213. Tsai
S, Shameli A, Yamanouchi J et al. Reversal of autoimmunity by boosting
memory-like autoregulatory T cells. Immunity 2010;32(4):568-580.
214. Pozzilli
P, Pitocco D, Visalli N et al. No effect of oral insulin on residual beta-cell
function in recent-onset type I diabetes (the IMDIAB VII). IMDIAB Group [In
Process Citation]. diabetol 2000;43(8):1000-1004.
215. Wilson
DM. Progress in the treatment of childhood diabetes mellitus and obesity. J
Pediatr Endocrinol Metab 2002;15 Suppl 2:745-749.
216. Skyler
JS, Krischer JP, Wolfsdorf J et al. Effects of oral insulin in relatives of
patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diab care
2005;28(5):1068-1076.
217. Lernmark
A, Agardh CD. Immunomodulation with human recombinant autoantigens. Trends
Immunol 2005;26(11):608-612.
218. Alleva
DG, Gaur A, Jin L et al. Immunological Characterization and Therapeutic
Activity of an Altered-Peptide Ligand, NBI-6024, Based on the Immunodominant
Type 1 Diabetes Autoantigen Insulin B-Chain (9-23) Peptide. diab
2002;51(7):2126-2134.
219. Pugliese
A. Peptide-based treatment for autoimmune diseases: learning how to handle a
double-edged sword. J Clin Invest 2003;111(9):1280-1282.
220. Ohneda
K, Ee H, German M. Regulation of insulin gene transcription. Semin Cell Dev
Biol 2000;11(4):227-233.
221. Throsby
M, Homo-Delarche F, Chevenne D, Goya R, Dardenne M, Pleau JM. Pancreatic
hormone expression in the murine thymus: localization in dendritic cells and
macrophages. Endocrinology 1998;139(5):2399-2406.
222. Sospedra
M, Ferrer-Francesch X, Dominguez O, Juan M, Foz-Sala M, Pujol-Borrell R.
Transcription of a broad range of self-antigens in human thymus suggests a role
for central mechanisms in tolerance toward peripheral antigens. J Immunol
1998;161(11):5918-5929.
223. Anderson
MS, Venanzi ES, Klein L et al. Projection of an immunological self shadow
within the thymus by the aire protein. Science 2002;298(5597):1395-1401.
224. Pugliese
A. Central and peripheral autoantigen presentation in immune tolerance.
Immunology 2004;111:138-146.
225. Vafiadis
P, Bennett ST, Todd JA et al. Insulin expression in human thymus is modulated
by INS VNTR alleles at the IDDM2 locus. Nat Genet 1997;15:289-292.
226. Pugliese
A, Zeller M, Fernandez A et al. The insulin gene is transcribed in the human
thymus and transcription levels correlate with allelic variation at the INS
VNTR-IDDM2 susceptibility locus for type I diabetes. Nat Genet
1997;15(3):293-297.
227. Todd
JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes
in humans and animal models. Immunity 2001;15(3):387-395.
228. Chentoufi
AA, Polychronakos C. Insulin expression levels in the thymus modulate
insulin-specific autoreactive T-cell tolerance: the mechanism by which the
IDDM2 locus may predispose to diabetes. diab 2002;51(5):1383-1390.
229. French
MB, Allison J, Cram DS et al. Transgenic expression of mouse proinsulin II
prevents diabetes in nonobese diabetic mice. diab 1996;46:34-39.
230. Pugliese
A, Diez J. Lymphoid organs contain diverse cells expressing self-molecules. Nat
Immunol 2002;3(4):335-336.
231. Gotter
J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human
thymus express a highly diverse selection of tissue-specific genes coloalized
in chromosomal clusters. J Exp Med 2004;199:155-166.
232. Chentoufi
AA, Palumbo M, Polychronakos C. Proinsulin expression by Hassall's corpuscles
in the mouse thymus. diab 2004;53(2):354-359.
233. Watanabe
N, Wang YH, Lee HK et al. Hassall's corpuscles instruct dendritic cells to
induce CD4+CD25+ regulatory T cells in human thymus. Nature
2005;436(7054):1181-1185.
234. Jaeckel
E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but
does not abolish type 1 diabetes. Nat Immunol 2004;5(10):1028-1035.
235. Kojima
H, Fujimiya M, Matsumura M, Nakahara T, Hara M, Chan L. Extrapancreatic insulin
producing cells in multiple organs in diabetes. 101[8], 2458-2463. 2004.
236. Kobayashi
M, Jasinski J, Liu E et al. Conserved T cell receptor alpha-chain induces
insulin autoantibodies. Proc Natl Acad Sci U S A 2008;105(29):10090-10094.
237. Homann
D, Eisenbarth GS. An immunologic homunculus for type 1 diabetes. J Clin Invest
2006;116(5):1212-1215.
238. Durinovic-Bello
I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype
associates with frequency and phenotype of the autoimmune response to
proinsulin. Genes Immun 2010.
239. Stadinski
BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T
cells recognize insulin bound to IAg7 in an unexpected, weakly binding
register. Proc Natl Acad Sci U S A 2010;107(24):10978-10983.
240. Levisetti
MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese
diabetic mice recognize a weak MHC-binding segment in more than one form. J
Immunol 2007;178(10):6051-6057.
241. Crawford
F, Stadinski B, Jin N et al. Specificity and detection of insulin-reactive CD4+
T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad
Sci U S A 2011.
242. Mohan
JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique
autoreactive T cells recognize insulin peptides generated within the islets of
Langerhans in autoimmune diabetes. Nat Immunol 2010;11(4):350-354.
243. Daniel
C, Weigmann B, Bronson R, von BH. Prevention of type 1 diabetes in mice by
tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med
2011;208(7):1501-1510.
244. Fan
Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of
insulin induces autoimmune diabetes. EMBO J 2009;28(18):2812-2824.
245. Daniel
C, Weigmann B, Bronson R, von BH. Prevention of type 1 diabetes in mice by
tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med
2011;208(7):1501-1510.
246. Lernmark
A. Glutamic acid decarboxylase--gene to antigen to disease. [Review] [85 refs].
J Intern Med 1996;240(5):259-277.
247. Kim
J, Richter W, Aanstoot HJ et al. Differential expression of GAD65 and GAD67 in
human, rat and mouse pancreatic islets. diab 1993;42:1799-1808.
248. Velloso
LA, Eizirik DL, Karlsson FA, Kampe O. Absence of autoantibodies against
glutamate decarboxylase (GAD) in the non-obese diabetic (NOD) mouse and low
expression of the enzyme in mouse islets. Clin Exp Immunol 1994;96(1):129-137.
249. Bieg
S, Seissler J, Herberg L, Northemann W, Scherbaum WA. GAD65 is recognized by
T-cells, but not by antibodies from NOD-mice. Autoimmunity 1994;17(3):189-194.
250. Kaufman
DL, Clare-Salzler M, Tian J et al. Spontaneous loss of T-cell tolerance to
glutamic acid decarboxylase in murine insulin-dependent diabetes [see
comments]. Nature 1993;366:69-72.
251. Tisch
R, Yang X-D, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to
glutamic acid decarboxylase correlates with insulitis in non-obese diabetic
mice. Nature 1993;366:72-75.
252. Quinn
A, McInerney B, Reich EP, Kim O, Jensen KP, Sercarz EE. Regulatory and effector
CD4 T cells in nonobese diabetic mice recognize overlapping determinants on
glutamic acid decarboxylase and use distinct V beta genes. J Immunol
2001;166(5):2982-2991.
253. Zechel
MA, Elliott JF, Atkinson MA, Singh B. Characterization of novel T-cell epitopes
on 65 kDa and 67 kDa glutamic acid decarboxylase relevant in autoimmune
responses in NOD mice. J Autoimmun 1998;11:83-95.
254. Chao
CC, McDevitt HO. Identification of immunogenic epitopes of GAD 65 presented by
Ag7 in non-obese diabetic mice. Immunogenetics 1997;46(1):29-34.
255. Videbaek
N, Harach S, Phillips J et al. An islet-homing NOD CD8+ cytotoxic T cell clone
recognizes GAD65 and causes insulitis. J Autoimmun 2003;20(2):97-109.
256. Atkinson
MA, Kaufman DL, Campbell L et al. Response of peripheral-blood mononuclear
cells to glutamate decarboxylase in insulin-dependent diabetes. Lancet
1992;339(8791):458-459.
257. Danke
NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T
cells in healthy and type 1 diabetic subjects. J Autoimmun 2005;25(4):303-311.
258. Ott
PA, Herzog BA, Quast S et al. Islet-cell antigen-reactive T cells show
different expansion rates and Th1/Th2 differentiation in type 1 diabetic
patients and healthy controls. Clin Immunol 2005;115(1):102-114.
259. Jaume
JC, Parry SL, Madec AM, Sonderstrup G, Baekkeskov S. Suppressive effect of
glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing
of T cell determinants located within the antibody epitope. J Immunol 2002;169(2):665-672.
260. Reijonen
H, Daniels TL, Lernmark A, Nepom GT. GAD65-specific autoantibodies enhance the
presentation of an immunodominant T-cell epitope from GAD65. diab
2000;49(10):1621-1626.
261. Liu
J, Purdy LE, Rabinovitch S, Jevnikar AM, Elliott JF. Major DQ8-restricted
T-cell epitopes for human GAD65 mapped using human CD4, DQA1*0301, DQB1*0302
transgenic IA(null) NOD mice. diab 1999;48(3):469-477.
262. Nepom
GT, Lippolis JD, White FM et al. Identification and modulation of a naturally
processed T cell epitope from the diabetes-associated autoantigen human
glutamic acid decarboxylase 65 (hGAD65). Proc Natl Acad Sci U S A
2001;98(4):1763-1768.
263. Ou
D, Mitchell LA, Metzger DL, Gillam S, Tingle AJ. Cross-reactive rubella virus
and glutamic acid decarboxylase (65 and 67) protein determinants recognised by
T cells of patients with type I diabetes mellitus. diabetol 2000;43(6):750-762.
264. Lohmann
T, Leslie RDG, Londei M. T cell clones to epitopes of glutamic acid
decarboxylase 65 raised from normal subjects and patients with
insulin-dependent diabetes. J Autoimmun 1996;9:385-389.
265. Schloot
NC, Roep BO, Wegmann DR, Yu L, Wang TB, Eisenbarth GS. T cell reactivity to
GAD65 peptide sequences shared with coxsackie virus protein in recent-onset
IDDM patients and control subjects. diabetol 1997;40(3):332-338.
266. Endl
J, Otto H, Jung G et al. Identification of naturally processed T cell epitopes
from glutamic acid decarboxylase presented in the context of HLA-DR alleles by
T lymphocytes of recent onset IDDM patients. J Clin Invest
1997;99(10):2405-2415.
267. Bach
JM, Otto H, Nepom GT et al. High affinity presentation of an autoantigenic
peptide in type I diabetes by an HLA class II protein encoded in a haplotype
protecting from disease. J Autoimmun 1997;10(4):375-386.
268. Boyton
RJ, Lohmann T, Londei M et al. Glutamic acid decarboxylase T lymphocyte
responses associated with susceptibility or resistance to type I diabetes:
analysis in disease discordant human twins, non-obese diabetic mice and HLA-DQ
transgenic mice. Int Immunol 1998;10(12):1765-1776.
269. Oling
V, Marttila J, Ilonen J et al. GAD65- and proinsulin-specific CD4+ T-cells
detected by MHC class II tetramers in peripheral blood of type 1 diabetes
patients and at-risk subjects. J Autoimmun 2005;25(3):235-243.
270. Hiemstra
HS, Schloot NC, van Veelen PA et al. Cytomegalovirus in autoimmunity: T cell
crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase.
Proc Natl Acad Sci U S A 2001;98(7):3988-3991.
271. Pak
CY, Eun H-M, McArthur RG, Yoon J-W. Association of cytomegalovirus infection
with autoimmune type I diabetes. Lancet 1988;2(8601):1-4.
272. Atkinson
MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular
immunity to a determinant common to glutamate decarboxylase and coxsackie virus
in insulin-dependent diabetes. J Clin Invest 1994;94(5):2125-2129.
273. Rudy
G, Stone N, Harrison LC et al. Similar peptides from two b cell autoantigens, proinsulin and glutamic acid
decarboxylase, stimulate T cells of individuals at risk for insulin-dependent
diabetes. Mol Med 1995;1(6):625-633.
274. Schloot
NC, Willemen SJ, Duinkerken G, Drijfhout JW, de Vries RR, Roep BO. Molecular
mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides
with sequence homology to Coxsackie or proinsulin peptides do not crossreact
with homologous counterpart. Hum Immunol 2001;62(4):299-309.
275. Panina-Bordignon
P, Lang R, Van Endert PM et al. Cytotoxic T cells specific for glutamic acid
decarboxylase in autoimmune diabetes. J Exp Med 1995;181(5):1923-1927.
276. Nepom
G, Quinn A, Sercarz E, Wilson DB. How important is GAD in the etiology of
spontaneous disease in human and murine type 1 diabetes? J Autoimmun
2003;20(3):193-194.
277. Yoon
JW, Yoon CS, Lim HW et al. Control of autoimmune diabetes in NOD mice by GAD
expression or suppression in beta cells. Science 1999;284(5417):1183-1187.
278. Jaeckel
E, Klein L, Martin-Orozco N, von Boehmer H. Normal incidence of diabetes in NOD
mice tolerant to glutamic acid decarboxylase. J Exp Med 2003;197(12):1635-1644.
279. Petersen
JS, Karlsen AE, Markholst H, Worsaae A, Dyrberg T, Michelsen B. Neonatal
tolerization with glutamic acid decarboxylase but not with bovine serum albumin
delays the onset of diabetes in NOD mice. diab 1994;43(12):1478-1484.
280. Tian
J, Atkinson M, Clare-Salzer M et al. Nasal administration of glutamate
decarboxylase (GAD65) peptides induces Th2 responses and prevents murine
insulin dependent diabetes. J Exp Med 1996;183:1561-1567.
281. Tian
J, Clare-Salzler M, Herschenfeld A et al. Modulating autoimmune responses to
GAD inhibits disease progression and prolongs islet graft survival in
diabetes-prone mice. Nat Med 1996;2(12):1348-1353.
282. Ramiya
VK, Shang XZ, Wasserfall CH, Maclaren NK. Effect of oral and intravenous
insulin and glutamic acid decarboxylase in NOD mice. Autoimmunity
1997;26(3):139-151.
283. Han
G, Li Y, Wang J et al. Active tolerance induction and prevention of autoimmune
diabetes by immunogene therapy using recombinant adenoassociated virus
expressing glutamic acid decarboxylase 65 peptide GAD(500-585). J Immunol
2005;174(8):4516-4524.
284. Elliott
JF, Qin HY, Bhatti S et al. Immunization with the larger isoform of mouse
glutamic acid decarboxylase (GAD67) prevents autoimmune diabetes in NOD mice.
diab 1994;43(12):1494-1499.
285. Bridgett
M, Cetkovic-Cvrlje M, O'Rourke R et al. Differential protection in two
transgenic lines of NOD/Lt mice hyperexpressing the autoantigen GAD65 in
pancreatic beta-cells. diab 1998;47(12):1848-1856.
286. Geng
L, Solimena M, Flavell RA, Sherwin RS, Hayday AC. Widespread expression of an
autoantigen-GAD65 transgene does not tolerize non-obese diabetic mice and can
exacerbate disease. Proc Natl Acad Sci USA 1998;95(17):10055-10060.
287. Jasinski
JM, Yu L, Nakayama M et al. Transgenic insulin (B:9-23) T-cell receptor mice
develop autoimmune diabetes dependent upon RAG genotype, H-2g7 homozygosity,
and insulin 2 gene knockout. Diabetes 2006;55(7):1978-1984.
288. Kim
SK, Tarbell KV, Sanna M et al. Prevention of type I diabetes transfer by
glutamic acid decarboxylase 65 peptide 206-220-specific T cells. Proc Natl Acad
Sci U S A 2004;101(39):14204-14209.
289. Gebe
JA, Unrath KA, Yue BB, Miyake T, Falk BA, Nepom GT. Autoreactive human T-cell
receptor initiates insulitis and impaired glucose tolerance in HLA DR4
transgenic mice. J Autoimmun 2008;30(4):197-206.
290. Wherrett
DK, Bundy B, Becker DJ et al. Antigen-based therapy with glutamic acid
decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a
randomised double-blind trial. Lancet 2011;378(9788):319-327.
291. Leslie
RD, Atkinson MA, Notkins AL. Autoantigens IA-2 and GAD in Type I
(insulin-dependent) diabetes. diabetol 1999;42(1):3-14.
292. Andersen
JN, Jansen PG, Echwald SM et al. A genomic perspective on protein tyrosine
phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J
2004;18(1):8-30.
293. Christie
MR, Tun RYM, Lo SSS et al. Antibodies to GAD and tryptic fragments of islet 64K
antigen as distinct markers for development of IDDM: Studies with identical
twins. diab 1992;41:782-787.
294. Hawkes
CJ, Wasmeier C, Christie MR, Hutton JC.
Identification of the 37-kDa antigen in IDDM as a tyrosine
phosphatase-like protein (phogrin) related to IA-2. diab 1996;45(9):1187-1192.
295. Bonifacio
E, Lampasona V, Genovese S, Ferrari M, Bosi E. Identification of protein
tyrosine phosphatase-like IA2 (islet call antigen 512) as the insulin-dependent
diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. J
Immunol 1995;155:5419-5426.
296. Kawasaki
E, Yu L, Rewers MJ, Hutton JC, Eisenbarth GS. Definition of multiple
ICA512/phogrin autoantibody epitopes and detection of intramolecular epitope
spreading in relatives of patients with type 1 diabetes. diab 1998;47:733-742.
297. Rabin
DU, Pleasic SM, Shapiro JA et al. Islet cell antigen 512 is a diabetes-specific
islet autoantigen related to protein tyrosine phosphatases. J Immunol
1994;152(6):3183-3188.
298. Payton
MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-Mr
tryptic fragments of islet antigens in insulin-dependent diabetes to the
protein tyrosine phosphatase-like molecule IA-2 (ICA512). J Clin Invest
1995;96:1506-1511.
299. LU
J, Li Q, Xie H et al. Identification of a second transmembrane protein tyrosine
phosphatase, IA-2b, as an
autoantigen in insulin-dependent diabetes mellitus: precursor of the 37-kDa
tryptic fragment. Proc Natl Acad Sci USA 1996;93:2307-2311.
300. Christie
MR, Genovese S, Cassidy D et al. Antibodies to islet 37k antigen, but not to
glutamate decarboxylase, discriminate rapid progression to IDDM in endocrine
autoimmunity. diab 1994;43(10):1254-1259.
301. Achenbach
P, Warncke K, Reiter J et al. Stratification of type 1 diabetes risk on the
basis of islet autoantibody characteristics. diab 2004;53(2):384-392.
302. Christie
MR, Vohra G, Champagne P, Daneman D, Delovitch TL. Distinct antibody
specificities to a 64-kD islet cell antigen in Type 1 diabetes as revealed by
trypsin treatment. J Exp Med 1990.
303. Lan
MS, LU J, Goto Y, Notkins AL. Molecular cloning and identification of a
receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA
Cell Biol 1994;13(5):505-514.
304. Rabin
DU, Pleasic SM, Palmer-Crocker R, Shapiro JA. Cloning and expression of
IDDM-specific human autoantigens. diab 1992;41(2):183-186.
305. Wasmeier
C, Hutton JC. Molecular cloning of phogrin, a protein-tyrosine phosphatase
homologue localized to insulin secretory granule membranes. J Biol Chem
1996;271(30):18161-18170.
306. Lan
MS, Wasserfall C, Maclaren NK, Notkins AL. IA-2, a transmembrane protein of the
protein tyrosine phosphatase family, is a major autoantigen in
insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A
1996;93(13):6367-6370.
307. Smith
PD, Barker KT, Wang J, Lu YJ, Shipley J, Crompton MR. ICAAR, a novel member of
a new family of transmembrane, tyrosine phosphatase-like proteins. Biochem
Biophys Res Commun 1996;229(2):402-411.
308. Cui
L, Yu WP, DeAizpurua HJ, Schmidli RS, Pallen CJ. Cloning and characterization
of islet cell antigen-related protein-tyrosine phosphatase (PTP), a novel
receptor-like PTP and autoantigen in insulin-dependent diabetes. J Biol Chem
1996;271(40):24817-24823.
309. Kawasaki
E, Hutton JC, Eisenbarth GS. Molecular cloning and characterization of the
human transmembrane protein tyrosine phosphatase homologue, phogrin, an
autoantigen of type 1 diabetes. Biochem Biophys Res Commun 1996;227:440-447.
310. Li
Q, Borovitskaya AE, DeSilva MG et al. Autoantigens in insulin-dependent
diabetes mellitus: molecular cloning and characterization of human IA-2 beta.
Proc Assoc Am Physicians 1997;109(4):429-439.
311. Solimena
M, Dirkx R, Jr., Hermel JM et al. ICA 512, an autoantigen of type I diabetes,
is an intrinsic membrane protein of neurosecretory granules. EMBO J
1996;15(9):2102-2114.
312. Diez
J, Park Y, Zeller M et al. Differential splicing of the IA-2 mRNA in pancreas
and lymphoid organs as a permissive genetic mechanism for autoimmunity against
the IA-2 type 1 diabetes autoantigen. diab 2001;50(4):895-900.
313. Genovese
S, Bonfanti R, Bazzigaluppi E et al. Association of IA-2 autoantibodies with
HLA DR4 phenotypes in IDDM. diabetol 1996;39(10):1223-1226.
314. Bearzatto
M, Naserke H, Piquer S et al. Two distinctly HLA-associated contiguous linear
epitopes uniquely expressed within the islet antigen 2 molecule are major
autoantibody epitopes of the diabetes-specific tyrosine phosphatase-like
protein autoantigens. Journal of Immunology 2002;168(8):4202-4208.
315. Lampasona
V, Bearzatto M, Genovese S, Bosi E, Ferrari M, Bonifacio E. Autoantibodies in
insulin-dependent diabetes recognize distinct cytoplasmic domain of the protein
tyrosine phosphatase-like IA-2 autoantigen. J Immunol 1996;157:2707-2711.
316. Hermel
JM, Dirkx R, Jr., Solimena M. Post-translational modifications of ICA512, a
receptor tyrosine phosphatase-like protein of secretory granules. Eur J
Neurosci 1999;11(8):2609-2620.
317. Ort
T, Voronov S, Guo J et al. Dephosphorylation of beta2-syntrophin and
Ca2+/mu-calpain-mediated cleavage of ICA512 upon stimulation of insulin
secretion. EMBO J 2001;20(15):4013-4023.
318. Chiang
M-K, Flanagan JG. PTP-NP, a new member of the receptor protein tyrosine phosphatase
family, implicated in development of nervous system and pancreatic endocrine
cells. Development 1996;122:2239-2250.
319. Drake
PG, Peters GH, Andersen HS, Hendriks W, Moller NP. A novel strategy for the
development of selective active-site inhibitors of the protein tyrosine
phosphatase-like proteins islet-cell antigen 512 (IA-2) and phogrin (IA-2beta).
Biochem J 2003;373(Pt 2):393-401.
320. Ort
T, Maksimova E, Dirkx R et al. The receptor tyrosine phosphatase-like protein
ICA512 binds the PDZ domains of beta2-syntrophin and nNOS in pancreatic
beta-cells. Eur J Cell Biol 2000;79(9):621-630.
321. Saeki
K, Zhu M, Kubosaki A, Xie J, Lan MS, Notkins AL. Targeted disruption of the
protein tyrosine phosphatase-like molecule IA-2 results in alterations in
glucose tolerance tests and insulin secretion. diab 2002;51(6):1842-1850.
322. Koczwara
K, Schenker M, Schmid S, Kredel K, Ziegler AG, Bonifacio E. Characterization of
antibody responses to endogenous and exogenous antigen in the nonobese diabetic
mouse. Clin Immunol 2003;106(2):155-162.
323. Durinovic-Bello
I, Hummel M, Ziegler AG. Cellular immune response to diverse islet cell
antigens in IDDM. diab 1996;45(6):795-800.
324. Dotta
F, Dionisi S, Viglietta V et al. T-cell mediated autoimmunity to the
insulinoma-associated protein 2 islet tyrosine phosphatase in type 1 diabetes
mellitus. Eur J Endocrinol 1999;141(3):272-278.
325. Kelemen
K, Crawford ML, Gill RG, Hutton JC, Wegmann D. Cellular immune response to
phogrin in the NOD mouse: cloned T-cells cause destruction of islet
transplants. diab 1999;48(8):1529-1534.
326. Herzog
BA, Ott PA, Dittrich MT et al. Increased in vivo frequency of IA-2
peptide-reactive IFNgamma+/IL-4- T cells in type 1 diabetic subjects. J
Autoimmun 2004;23(1):45-54.
327. Muntasell
A, Carrascal M, Alvarez I et al. Dissection of the HLA-DR4 peptide repertoire
in endocrine epithelial cells: strong influence of invariant chain and HLA-DM
expression on the nature of ligands. J Immunol 2004;173(2):1085-1093.
328. Kelemen
K, Wegmann DR, Hutton JC. T-cell epitope analysis on the autoantigen phogrin
(IA-2beta) in the nonobese diabetic mouse. diab 2001;50(8):1729-1734.
329. Achenbach
P, Kelemen K, Wegmann DR, Hutton JC. Spontaneous peripheral T-cell responses to
the IA-2beta (phogrin) autoantigen in young nonobese diabetic mice. J Autoimmun
2002;19(3):111-116.
330. Kelemen
K, Gottlieb PA, Putnam AL, Davidson HW, Wegmann DR, Hutton JC.
HLA-DQ8-associated T cell responses to the diabetes autoantigen phogrin
(IA-2beta) in human prediabetes. J Immunol 2004;172(6):3955-3962.
331. Kudva
YC, Deng YJ, Govindarajan R et al. HLA-DQ8 transgenic and NOD mice recognize
different epitopes within the cytoplasmic region of the tyrosine
phosphatase-like molecule, IA-2. Hum Immunol 2001;62(10):1099-1105.
332. Trembleau
S, Penna G, Gregori S, Magistrelli G, Isacchi A, Adorini L. Early Th1 response
in unprimed nonobese diabetic mice to the tyrosine phosphatase-like
insulinoma-associated protein 2, an autoantigen in type 1 diabetes. J Immunol 2000;165(12):6748-6755.
333. Honeyman
MC, Stone NL, Harrison LC. T-cell epitopes in type 1 diabetes autoantigen
tyrosine phosphatase IA-2: Potential for mimcry with rotavirus and other
environmental agents. Mol Med 1998;4:231-239.
334. Peakman
M, Stevens EJ, Lohmann T et al. Naturally processed and presented epitopes of
the islet cell autoantigen IA-2 eluted from HLA-DR4 [see comments]. J Clin
Invest 1999;104(10):1449-1457.
335. Takahashi
K, Honeyman MC, Harrison LC. Cytotoxic T cells to an epitope in the islet
autoantigen IA-2 are not disease-specific. Clin Immunol 2001;99(3):360-364.
336. Cai
T, Krause MW, Odenwald WF, Toyama R, Notkins AL. The IA-2 gene family: homologs
in Caenorhabditis elegans, Drosophila and zebrafish. diabetol 2001;44(1):81-88.
337. Zahn
TR, Macmorris MA, Dong W, Day R, Hutton JC. IDA-1, a Caenorhabditis elegans
homolog of the diabetic autoantigens IA-2 and phogrin, is expressed in
peptidergic neurons in the worm. J Comp Neurol 2001;429(1):127-143.
338. Park
YS, Kawasaki E, Kelemme K et al. Humoral autoreactivity to an alternative
spliced variant of ICA512/IA2 in type 1 diabetes. diabetol 2000;50:895-900.
339. Ebert
DH, Bischof LJ, Streeper RS et al. Structure and promoter activity of an
islet-specific glucose-6- phosphatase catalytic subunit-related gene. diab
1999;48(3):543-551.
340. Bischof
LJ, Martin CC, Svitek CA et al. Characterization of the mouse islet-specific
glucose-6-phosphatase catalytic subunit-related protein gene promoter by in
situ footprinting: correlation with fusion gene expression in the islet-derived
betaTC-3 and hamster insulinoma tumor cell lines. diab 2001;50(3):502-514.
341. Arden
SD, Zahn T, Steegers S et al. Molecular cloning of a pancreatic islet-specific
glucose-6-phosphatase catalytic subunit-related protein. diab
1999;48(3):531-542.
342. Hutton
JC, Eisenbarth GS. A pancreatic {beta}-cell-specific homolog of
glucose-6-phosphatase emerges as a major target of cell-mediated autoimmunity
in diabetes. Proc Natl Acad Sci U S A 2003;100:8626-8628.
343. Shieh
JJ, Pan CJ, Mansfield BC, Chou JY. The islet-specific
glucose-6-phosphatase-related protein, implicated in diabetes, is a
glycoprotein embedded in the endoplasmic reticulum membrane. FEBS Lett
2004;562(1-3):160-164.
344. DiLorenzo
TP, Graser RT, Ono T et al. Major histocompatibility complex class I-restricted
T cells are required for all but the end stages of diabetes development in
nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene
rearrangement. Proc Natl Acad Sci USA 1998;95(21):12538-12543.
345. Verdaguer
J, Yoon J-W, Anderson B et al. Acceleration of spontaneous diabetes in TCR-b-transgenic nonobese diabetic mice by b-cell cytotoxic CD8+ T cells expressing
identical endognous TCR-a chains. J
Immunol 1996;157:4726-4735.
346. Verdaguer
J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous
autoimmune diabetes in monoclonal T cell nonobese diabetic mice. Journal of
Experimental Medicine 1997;186(10):1663-1676.
347. Tsai
S, Shameli A, Santamaria P. CD8+ T cells in type 1 diabetes. Adv Immunol
2008;100:79-124.
348. Anderson
B, Park BJ, Verdaguer J, Amrani A, Santamaria P. Prevalent CD8+ T
cell response against one peptide/MHC complex in autoimmune diabetes. Proc Natl
Acad Sci U S A 1999;96(16):9311-9316.
349. Amrani
A, Serra P, Yamanouchi J et al. Expansion of the antigenic repertoire of a
single T cell receptor upon T cell activation. J Immunol 2001;167(2):655-666.
350. Trudeau
JD, Kelly-Smith C, Verchere CB et al. Prediction of spontaneous autoimmune
diabetes in NOD mice by quantification of autoreactive T cells in peripheral
blood. J Clin Invest 2003;111(2):217-223.
351. Moore
A, Grimm J, Han B, Santamaria P. Tracking the recruitment of diabetogenic CD8+
T-cells to the pancreas in real time. diab 2004;53(6):1459-1466.
352. Han
B, Serra P, Yamanouchi J et al. Developmental control of CD8 T cell-avidity
maturation in autoimmune diabetes. J Clin Invest 2005;115(7):1879-1887.
353. Han
B, Serra P, Amrani A et al. Prevention of diabetes by manipulation of anti-IGRP
autoimmunity: high efficiency of a low-affinity peptide. Nat Med
2005;11(6):645-652.
354. Jarchum
I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP
epitopes targeted in type 1 diabetes patients. Clin Immunol
2008;127(3):359-365.
355. Wenzlau
JM, Juhl K, Yu L et al. The cation efflux transporter ZnT8 (Slc30A8) is a major
autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A
2007;104(43):17040-17045.
356. Wenzlau
JM, Moua O, Sarkar SA et al. SlC30A8 is a major target of humoral autoimmunity
in type 1 diabetes and a predictive marker in prediabetes. Ann N Y Acad Sci
2008;1150:256-259.
357. De
GJ, Asanghanwa M, Nouthe B et al. Predictive power of screening for antibodies
against insulinoma-associated protein 2 beta (IA-2beta) and zinc transporter-8
to select first-degree relatives of type 1 diabetic patients with risk of rapid
progression to clinical onset of the disease: implications for prevention
trials. diabetol 2009.
358. Yu
L, Liu Y, Miao D et al. Triple chimeric islet autoantigen IA2-ZnT8WR to
facilitate islet autoantibody determination. J Immunol Methods
2010;353(1-2):20-23.
359. Wenzlau
JM, Moua O, Liu Y, Eisenbarth GS, Hutton JC, Davidson HW. Identification of a
major humoral epitope in Slc30A8 (ZnT8). Ann N Y Acad Sci 2008;1150:252-255.
360. Wenzlau
JM, Walter M, Gardner TJ et al. Kinetics of the Post-Onset Decline in Zinc
Transporter 8 Autoantibodies in Type 1 Diabetic Human Subjects. J Clin
Endocrinol Metab 2010.
361. Poulin
M, Haskins K. Induction of diabetes in nonobese diabetic mice by Th2 T cell
clones from a TCR transgenic mouse. J Immunol 2000;164(6):3072-3078.
362. Wucherpfennig
KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide-MHC
recognition by self-reactive T cell receptors. Curr Opin Immunol
2009;21(6):590-595.
363. Molberg
O, McAdam S, Lundin KE et al. T cells from celiac disease lesions recognize
gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur
J Immunol 2001;31(5):1317-1323.
364. Pietropaolo
M, Castano L, Babu S et al. Islet cell autoantigen 69 kDa (ICA69): molecular
cloning and characterization of a novel diabetes associated autoantigen. J Clin
Invest 1993;92:359-371.
365. Spitzenberger
F, Pietropaolo S, Verkade P et al. Islet cell autoantigen of 69 kDa is an
arfaptin-related protein associated with the Golgi complex of insulinoma INS-1
cells. J Biol Chem 2003;278(28):26166-26173.
366. Mally
MI, Cirulli V, Hayek A, Otonkoski T. ICA69 is expressed equally in the human
endocrine and exocrine pancreas. diabetol 1996;39:474-480.
367. Stassi
G, Schloot N, Pietropaolo M. Islet cell autoantigen 69 kDa (ICA69) is
preferentially expressed in the human islets of Langerhans than exocrine
pancreas. diabetol 1997;40:120-121.
368. Friday
RP, Pietropaolo SL, Profozich J, Trucco M, Pietropaolo M. Alternative core
promoters regulate tissue-specific transcription from the autoimmune
diabetes-related ICA1 (ICA69) gene locus. J Biol Chem 2003;278(2):853-863.
369. Mathews
CE, Pietropaolo SL, Pietropaolo M. Reduced thymic expression of islet antigen
contributes to loss of self-tolerance. Ann N Y Acad Sci 2003;1005:412-417.
370. Winer
S, Astsaturov I, Gaedigk R et al. ICA69(null) nonobese diabetic mice develop
diabetes, but resist disease acceleration by cyclophosphamide. J Immunol
2002;168(1):475-482.
371. Roep
BO, Duinkerken G, Schreuder GMT, Kolb H, De Vries RRP, Martin S. HLA-associated
inverse correlation between T cell and antibody responsiveness to islet
autoantigen in recent-onset insulin-dependent diabetes mellitus. Eur J Immunol
1996;26:1285-1289.
372. Miyazaki
I, Cheung RK, Gaedigk R et al. T cell activation and anergy to islet cell
antigen in type I diabetes. J Immunol 1995;154(3):1461-1469.
373. Karges
W, Hammond-McKibben D, Gaedigk R, Shibuya N, Cheung R, Dosch HM. Loss of
self-tolerance to ICA69 in nonobese diabetic mice. diab 1997;46(10):1548-1556.
374. Karjalainen
J, Martin JM, Knip M et al. A bovine albumin peptide as a possible trigger of
insulin- dependent diabetes mellitus. N Engl J Med 1992;327(5):302-307.
375. Norris
J, Pietropaolo M. A bovine albumin peptide as a possible trigger of
insulin-dependent diabetes mellitus. J Endocrinol Invest 1994;17(7):565-572.
376. Karlsson
MG, Ludvigsson J. The ABBOS-peptide from bovine serum albumin causes an
IFN-gamma and IL-4 mRNA response in lymphocytes from children with recent onset
of type 1 diabetes. Diabetes Res Clin Pract 2000;47(3):199-207.
377. Dosch
H, Cheung RK, Karges W, Pietropaolo M, Becker DJ. Persistent T cell anergy in
human type 1 diabetes. J Immunol 1999;163(12):6933-6940.
378. Steiner
DF. The proprotein convertases. Curr Opin Chem Biol 1998;2(1):31-39.
379. Davidson
HW. (Pro)Insulin processing: a historical perspective. Cell Biochem Biophys
2004;40(3 Suppl):143-158.
380. Davidson
HW, Hutton JC. The insulin-secretory-granule carboxypeptidase H. Purification
and demonstration of involvement in proinsulin processing. Biochem J
1987;245(2):575-582.
381. Guest
PC, Pipeleers D, Rossier J, Rhodes CJ, Hutton JC. Co-secretion of
carboxypeptidase H and insulin from isolated rat islets of Langerhans. Biochem
J 1989;264(2):503-508.
382. Hull
RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in
the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab
2004;89(8):3629-3643.
383. Betsholtz
C, Svensson V, Rorsman F et al. Islet amyloid polypeptide (IAPP):cDNA cloning
and identification of an amyloidogenic region associated with the
species-specific occurrence of age-related diabetes mellitus. Exp Cell Res
1989;183(2):484-493.
384. Sanke
T, Bell GI, Sample C, Rubenstein AH, Steiner DF. An islet amyloid peptide is
derived from an 89-amino acid precursor by proteolytic processing. J Biol Chem
1988;263(33):17243-17246.
385. Marzban
L, Trigo-Gonzalez G, Zhu X et al. Role of beta-cell prohormone convertase
(PC)1/3 in processing of pro-islet amyloid polypeptide. diab
2004;53(1):141-148.
386. Panagiotopoulos
C, Qin H, Tan R, Verchere CB. Identification of a beta-cell-specific HLA class
I restricted epitope in type 1 diabetes. diab 2003;52(11):2647-2651.
387. Dallas-Pedretti
A, McDuffie M, Haskins K. A diabetes-associated T cell autoantigen maps to a
telomeric locus on mouse chromosome 6. Proc Natl Acad Sci USA 1995;92:1386.
388. Res
P, Thole J, de VR. Heat-shock proteins and autoimmunity in humans. Springer
Semin Immunopathol 1991;13(1):81-98.
389. Brudzynski
K, Martinez V. Synaptophysin-containing microvesicles transport heat-shock
protein hsp60 in insulin-secreting beta cells. Cytotechnology 1993;11(1):23-33.
390. Tsan
MF, Gao B. Cytokine function of heat shock proteins. Am J Physiol Cell Physiol
2004;286(4):C739-C744.
391. Panayi
GS, Corrigall VM, Henderson B. Stress cytokines: pivotal proteins in immune
regulatory networks; Opinion. Curr Opin Immunol 2004;16(4):531-534.
392. Elias
D, Markovits D, Reshef T, Zee R, Cohen IR. Induction and therapy of autoimmune
diabetes in the non-obese diabetic (NOD/Lt) mouse by a 65-kDa heat shock
protein. Proc Natl Acad Sci USA 1990;87:1576-1580.
393. Shimada
A, Charlton B, Rohane P, Taylor-Edwards C, Fathman CG. Immune regulation in
type 1 diabetes. J Autoimmun 1996;9(2):263-269.
394. Elias
D, Cohen IR. Treatment of autoimmune diabetes and insulitis in NOD mice with
heat shock protein 60 peptide p277. diab 1995;44(9):1132-1138.
395. Elias
D, Cohen IR. Peptide therapy for diabetes in NOD mice. Lancet
1994;343(8899):704-706.
396. Bowman
M, Atkinson MA. Heat shock protein therapy fails to prevent diabetes in NOD
mice. diabetol 2002;45(9):1350-1351.
397. Ablamunits
V, Elias D, Cohen IR. The pathogenicity of islet-infiltrating lymphocytes in
the non-obese diabetic (NOD) mouse. Clin Exp Immunol 1999;115(2):260-267.
398. Elias
D, Cohen IR. The hsp60 peptide p277 arrests the autoimmune diabetes induced by
the toxin streptozotocin. diab 1996;45(9):1168-1172.
399. Feili-Hariri
M, Frantz MO, Morel PA. Prevention of diabetes in the NOD mouse by a Th1 clone
specific for a hsp60 peptide. J Autoimmun 2000;14(2):133-142.
400. Tikochinski
Y, Elias D, Steeg C et al. A shared TCR CDR3 sequence in NOD mouse autoimmune
diabetes. Int Immunol 1999;11(6):951-956.
401. Dosch
HM, Becker DJ. Measurement of T-cell autoreactivity in autoimmune diabetes
[letter] [In Process Citation]. diabetol 2000;43(3):386-387.
402. Kallmann
BA, Lampeter EF, Hanifi-Moghaddam P, Hawa M, Leslie RD, Kolb H. Cytokine
secretion patterns in twins discordant for type I diabetes. diabetol
1999;42(9):1080-1085.
403. Abulafia-Lapid
R, Gillis D, Yosef O, Atlan H, Cohen IR. T Cells and autoantibodies to human
HSP70 in Type 1 diabetes in children. J Autoimmun 2003;20(4):313-321.
404. Raz
I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR. Beta-cell function in
new-onset type 1 diabetes and immunomodulation with a heat-shock protein
peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet
2001;358(9295):1749-1753.
405. Hanninen
A, Hamilton-Williams E, Kurts C. Development of new strategies to prevent type
1 diabetes: the role of animal models. Ann Med 2003;35(8):546-563.
406. Raz
I, Avron A, Tamir M et al. Treatment of new-onset type 1 diabetes with peptide
DiaPep277 is safe and associated with preserved beta-cell function: extension
of a randomized, double-blind, phase II trial. Diabetes Metab Res Rev
2007;23(4):292-298.
407. Gelber
C, Paborsky L, Singer S et al. Isolation of nonobese diabetic mouse T-cells
that recognize novel autoantigens involved in the early events of diabetes.
diab 1994;43:33-39.
408. Santamaria
P, Utsugi T, Park B-J, Averill N, Kawazu S, Yoon J-W. Beta-cell-cytotoxic CD8+
T cells from nonobese diabetic mice use highly homologous T cell receptor a-chain CDR3 sequences. J Immunol 1995;154:2494-2503.
409. DiLorenzo
TP, Lieberman SM, Takaki T et al. During the early prediabetic period in NOD
mice, the pathogenic CD8(+) T-cell population comprises multiple antigenic
specificities. Clin Immunol 2002;105(3):332-341.
410. Yu
L, Rewers M, Gianani R et al. Anti-islet autoantibodies develop sequentially
rather than simultaneously. J Clin Endocrinol Metab 1996;81(12):4264-4267.
411. Palmer
JP, Asplin CM, Clemons P et al. Insulin antibodies in insulin-dependent
diabetics before insulin treatment. Science 1983;222(4630):1337-1339.
412. Castano
L, Eisenbarth GS. Type I diabetes: a chronic autoimmune disease of man, mouse,
and rat. Ann Rev Immunol 1990;8:647-679.
413. Wolfe
T, Bot A, Hughes A et al. Endogenous expression levels of autoantigens
influence success or failure of DNA immunizations to prevent type 1 diabetes:
addition of IL-4 increases safety. Eur J Immunol 2002;32(1):113-121.
414. Greiner
DL, Rossini AA, Mordes JP. Translating data from animal models into methods for
preventing human autoimmune diabetes mellitus: caveat emptor and primum non
nocere. Clin Immunol 2001;100(2):134-143.
415. Shimada
A, Charlton B, Taylor-Edwards C, Fathman CG. b-cell
destruction may be a late consequence of the autoimmune process in nonobese
diabetic mice. diab 1996;45:1063-1067.
416. Sreenan
S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS. Increased b-cell proliferation and reduced mass before diabetes
onset in the nonobese diabetic mouse. diab 1999;48:989-996.
417. Sherry
NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of
autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes.
Diabetes 2006;55(12):3238-3245.
418. Wicker
LS, Todd JA, Peterson LB. Genetic control of autoimmune diabetes in the NOD
mouse. Ann Rev Immunol 1995;13:179-200.
419. Ott
PA, Dittrich MT, Herzog BA et al. T cells recognize multiple GAD65 and
proinsulin epitopes in human type 1 diabetes, suggesting determinant spreading.
J Clin Immunol 2004;24(4):327-339.
420. Ueda
H, Howson JM, Esposito L et al. Association of the T-cell regulatory gene CTLA4
with susceptibility to autoimmune disease. Nature 2003;423(6939):506-511.
421. You
S, Belghith M, Cobbold S et al. Autoimmune diabetes onset results from
qualitative rather than quantitative age-dependent changes in pathogenic
T-cells. diab 2005;54(5):1415-1422.
422. Boitard
C, Yasunami R, Dardenne M, Bach JF. T cell-mediated inhibition of the transfer
of autoimmune diabetes in NOD mice. J Exp Med 1989;169(5):1669-1680.
423. Pop
SM, Wong CP, Culton DA, Clarke SH, Tisch R. Single cell analysis shows
decreasing FoxP3 and TGFbeta1 coexpressing CD4+CD25+ regulatory T cells during
autoimmune diabetes. J Exp Med 2005;201(8):1333-1346.
424. Kolb
H. Benign versus destructive insulitis. Diabetes Metab Rev 1997;13(3):139-146.
425. Liblau
RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of
organ-specific autoimmune diseases [see comments]. Immunol Today
1995;16(1):34-38.
426. Katz
JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes.
Science 1995;268:1185-1188.
427. Sarukhan
A, Lanoue A, Franzke A, Brousse N, Buer J, von BH. Changes in function of
antigen-specific lymphocytes correlating with progression towards diabetes in a
transgenic model. EMBO J 1998;17(1):71-80.
428. Nagata
S, Golstein P. The Fas death factor. Science 1995;267(5203):1449-1456.
429. Jun
HS, Santamaria P, Lim HW, Zhang ML, Yoon JW. Absolute requirement of
macrophages for the development and activation of beta-cell cytotoxic CD8+
T-cells in T-cell receptor transgenic NOD mice. diab 1999;48(1):34-42.
430. Hamilton-Williams
EE, Palmer SE, Charlton B, Slattery RM. Beta cell MHC class I is a late
requirement for diabetes. Proc Natl Acad Sci U S A 2003;100(11):6688-6693.
431. Eizirik
DL, Mandrup-Poulsen T. A choice of death - the signal-transduction of immune-mediated
beta-cell apoptosis. diabetol 2001;44(12):2115-2133.
432. Mandrup-Poulsen
T. Beta cell death and protection. Ann N Y Acad Sci 2003;1005:32-42.
433. Gianani
R, Campbell-Thompson M, Sarkar SA et al. Dimorphic histopathology of
long-standing childhood-onset diabetes. diabetol 2010.
434. Chatenoud
L. Anti-CD3 antibodies: towards clinical antigen-specific immunomodulation.
Curr Opin Pharmacol 2004;4(4):403-407.
435. Maki
T, Ichikawa T, Blanco R, Porter J. Long-term abrogation of autoimmune diabetes
in nonobese diabetic mice by immunotherapy with anti-lymphocyte serum. Proc
Natl Acad Sci U S A 1992;89(8):3434-3438.
436. Louvet
C, Szot GL, Lang J et al. Tyrosine kinase inhibitors reverse type 1 diabetes in
nonobese diabetic mice. Proc Natl Acad Sci U S A 2008.
437. Razavi
R, Chan Y, Afifiyan FN et al. TRPV1(+) Sensory Neurons Control beta Cell Stress
and Islet Inflammation in Autoimmune Diabetes. Cell 2006;127(6):1123-1135.
438. Keymeulen
B, Vandemeulebroucke E, Ziegler AG et al. Insulin needs after CD3-antibody
therapy in new-onset type 1 diabetes. N Engl J Med 2005;352(25):2598-2608.
439. Xue
S, Wasserfall CH, Parker M et al. Exendin-4 therapy in NOD mice with new-onset
diabetes increases regulatory T cell frequency. Ann N Y Acad Sci 2008;1150:152-156.
440. Faustman
DL. Immunotherapy on trial for new-onset type 1 diabetes. N Engl J Med
2008;359(18):1956-1958.
441. Fife
BT, Guleria I, Gubbels BM et al. Insulin-induced remission in new-onset NOD
mice is maintained by the PD-1-PD-L1 pathway. J Exp Med 2006;203(12):2737-2747.
442. Alegre
ML, Peterson LJ, Xu D et al. A non-activating "humanized" anti-CD3
monoclonal antibody retains immunosuppressive properties in vivo.
Transplantation 1994;57(11):1537-1543.
443. Herold
KC, Taylor L. Treatment of type 1 diabetes with anti-CD3 monoclonal antibody -
Induction of immune regulation? Immunologic Research 2003;28(2):141-150.
444. Bolt
S, Routledge E, Lloyd I et al. The generation of a humanized, non-mitogenic CD3
monoclonal antibody which retains in vitro immunosuppressive properties. Eur J
Immunol 1993;23(2):403-411.
445. Belghith
M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent
mechanisms mediate restoration of self-tolerance induced by antibodies to CD3
in overt autoimmune diabetes. Nat Med 2003;9(9):1202-1208.
446. Bisikirska
B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified
anti-CD3 mAb expands CD8 T cell population and induces CD8CD25 Tregs. J Clin
Invest 2005;115(10):2904-2913.
447. Karges
B, Durinovic-Bello I, Heinze E, Debatin KM, Boehm B, Karges W. Immunological
mechanisms associated with long-term remission of human type 1 diabetes.
Diabetes Metab Res Rev 2005;.
448. Ogawa
N, List JF, Habener JF, Maki T. Cure of overt diabetes in NOD mice by transient
treatment with anti-lymphocyte serum and exendin-4. diab 2004;53(7):1700-1705.
449. Mathis
D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature
2001;414(6865):792-798.
450. Thomas
HE, Kay TW. How beta cells die in type 1 diabetes. Curr Dir Autoimmun
2001;4:144-70.:144-170.
451. Signore
A, Annovazzi A, Gradini R, Liddi R, Ruberti G. Fas and Fas ligand-mediated
apoptosis and its role in autoimmune diabetes. [Review] [73 refs]. Diabetes
Metab Rev 1998;14(3):197-206.
452. Savinov
AY, Tcherepanov A, Green EA, Flavell RA, Chervonsky AV. Contribution of Fas to
diabetes development. Proc Natl Acad Sci U S A 2003;100(2):628-632.
453. Thomas
HE, Darwiche R, Corbett JA, Kay TW. Evidence that beta cell death in the
nonobese diabetic mouse is Fas independent. J Immunol 1999;163(3):1562-1569.
454. Lohmann
T, Halder T, Engler J et al. T cell reactivity to DR*. Exp Clin Endocrinol
Diabetes 1999;107(3):166-171.
455. Amrani
A, Verdaguer J, Anderson B, Utsugi T, Bou S, Santamaria P. Perforin-independent
beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic
nonobese diabetic mice. J Clin Invest 1999;103(8):1201-1209.
456. Suarez-Pinzon
WL, Mabley JG, Strynadka K, Power RF, Szabo C, Rabinovitch A. An inhibitor of
inducible nitric oxide synthase and scavenger of peroxynitrite prevents
diabetes development in NOD mice. J Autoimmun 2001;16(4):449-455.
457. Kolb
H, Kolb-Bachofen V. Nitric oxide in autoimmune disease: cytotoxic or regulatory
mediator? Immunol Today 1998;19(12):556-561.
458. Rabinovitch
A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM.
Therapeutic intervention by immunostimulation? diab 1994;43(5):613-621.
459. Gaur
U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members
of the TNF superfamily. Biochem Pharmacol 2003;66(8):1403-1408.
460. O'Reilly
LA, Hutchings PR, Crocker PR et al. Characterization of pancreatic islet cell
infiltrates in NOD mice: effect of cell transfer and transgene expression. Eur
J Immunol 1991;21(5):1171-1180.
461. Miller
BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets
are required for the transfer of diabetes in nonobese diabetic mice. J Immunol
1988;140(1):52-58.
462. Cooke
A, Phillips JM, Parish NM. Tolerogenic strategies to halt or prevent type 1
diabetes. Nat Immunol 2001;2(9):810-815.
463. Jansen
A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA.
Immunohistochemical characterization of monocytes-macrophages and dendritic
cells involved in the initiation of the insulitis and b-cell destruction in NOD mice. diab 1994;43:667-675.
464. Dilts
SM, Solvason N, Lafferty KJ. The role of CD4 and CD8 T cells in the development
of autoimmune diabetes. J Autoimmun 1999;13(3):285-290.
465. Shizuru
JA, Taylor-Edwards C, Banks BA, Gregory AK, Fathman GC. Immunotherapy of the
nonobese diabetic mouse: treatment with an antibody to T-helper lymphocytes.
Science 1988;240:659-662.
466. Wang
B, Gonzales A, Benoist C, Mathis D. The role of CD8+ T cells in the initiation
of insulin-dependent diabetes mellitus. Eur J Immunol 1996;26:1762-1769.
467. Abbas
AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature
1996;383(6603):787-793.
468. Mueller
R, Krahl T, Sarvetnick N. Pancreatic expression of interleukin-4 abrogates
insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med
1996;184(3):1093-1099.
469. Wang
B, Gonzalez A, Höglund P, Katz JD, Benoist C, Mathis D. Interleukin-4 deficiency
does not exacerbate disease in NOD mice. diab 1998;47:1207-1211.
470. Pakala
SV, Kurrer MO, Katz JD. T helper 2 (Th2) T cells induce acute pancreatitis and
diabetes in immune-compromised nonobese diabetic (NOD) mice. J Exp Med
1997;186(2):299-306.
471. Wogensen
L, Lee M-S, Sarvetnick N. Production of interleukin 10 by islet cells
accelerates immune-mediated destruction of b cells in
nonobese diabetic mice. J Exp Med 1994;179:1379-1384.
472. Izcue
A, Powrie F. Special regulatory T-cell review: Regulatory T cells and the
intestinal tract--patrolling the frontier. Immunology 2008;123(1):6-10.
473. Shevach
EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol
2002;2(6):389-400.
474. Bluestone
JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol
2003;3(3):253-257.
475. Malley
A, Baecher L. Inhibition of histamine and SRS-A from monkey lung tissue by
chlorophenesin. J Immunol 1971;107(2):586-588.
476. Horwitz
DA, Zheng SG, Gray JD. The role of the combination of IL-2 and TGF-beta or
IL-10 in the generation and function of CD4+ CD25+ and CD8+ regulatory T cell
subsets. J Leukoc Biol 2003;74(4):471-478.
477. van
der Vliet HJ, Molling JW, von Blomberg BM et al. The immunoregulatory role of
CD1d-restricted natural killer T cells in disease. Clin Immunol
2004;112(1):8-23.
478. Homann
D, von Herrath M. Regulatory T cells and type 1 diabetes. Clin Immunol
2004;112(3):202-209.
479. Gregori
S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T
cells in autoimmune diabetes development. Journal of Immunology
2003;171(8):4040-4047.
480. Esteban
LM, Tsoutsman T, Jordan MA et al. Genetic control of NKT cell numbers maps to
major diabetes and lupus loci. Journal of Immunology 2003;171(6):2873-2878.
481. Berzins
SP, Venanzi ES, Benoist C, Mathis D. T-Cell Compartments of Prediabetic NOD
Mice. diab 2003;52(2):327-334.
482. Lee
M, Koh JJ, Han SO, Ko KS, Ki SW. Prevention of autoimmune insulitis by delivery
of interleukin-4 plasmid using a soluble and biodegradable polymeric carrier.
Pharm Res 2002;19(3):246-249.
483. Maggi
E, Cosmi L, Liotta F, Romagnani P, Romagnani S, Annunziato F. Thymic regulatory
T cells. Autoimmun Rev 2005;4(8):579-586.
484. You
S, Slehoffer G, Barriot S, Bach JF, Chatenoud L. Unique role of CD4+CD62L+
regulatory T cells in the control of autoimmune diabetes in T cell receptor
transgenic mice. Proc Natl Acad Sci U S A 2004.
485. Sakaguchi
S. The origin of FOXP3-expressing CD4+ regulatory T cells: thymus or periphery.
J Clin Invest 2003;112(9):1310-1312.
486. Strobel
S. Oral tolerance, systemic immunoregulation, and autoimmunity. Ann N Y Acad
Sci 2002;958:47-58.
487. Weiner
HL. Induction and mechanism of action of transforming growth factor-beta-secreting
Th3 regulatory cells. Immunol Rev 2001;182:207-214.
488. Roncarolo
MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells.
Immunol Rev 2001;182:68-79.
489. Wakkach
A, Fournier N, Brun V, Breittmayer JP, Cottrez F, Groux H. Characterization of
dendritic cells that induce tolerance and T regulatory 1 cell differentiation
in vivo. Immunity 2003;18(5):605-617.
490. Dieckmann
D, Bruett CH, Ploettner H, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory,
contact-dependent T cells induce interleukin 10-producing, contact-independent
type 1-like regulatory T cells [corrected]. J Exp Med 2002;196(2):247-253.
491. Serra
P, Amrani A, Yamanouchi J et al. CD40 ligation releases immature dendritic
cells from the control of regulatory CD4+CD25+ T cells. Immunity
2003;19(6):877-889.
492. Feinberg
MB, Silvestri G. T(S) cells and immune tolerance induction: a regulatory
renaissance? Nat Immunol 2002;3(3):215-217.
493. Chess
L, Jiang H. Resurrecting CD8+ suppressor T cells. Nat Immunol
2004;5(5):469-471.
494. Horwitz
DA, Zheng SG, Gray JD, Wang JH, Ohtsuka K, Yamagiwa S. Regulatory T cells
generated ex vivo as an approach for the therapy of autoimmune disease. Semin
Immunol 2004;16(2):135-143.
495. Antony
PA, Restifo NP. Do CD4+ CD25+ immunoregulatory T cells hinder tumor
immunotherapy? J Immunother 2002;25(3):202-206.
496. Hammond
KJ, Kronenberg M. Natural killer T cells: natural or unnatural regulators of
autoimmunity? Curr Opin Immunol 2003;15(6):683-689.
497. Xiu
Y, Wong CP, Bouaziz JD et al. B lymphocyte depletion by CD20 monoclonal
antibody prevents diabetes in nonobese diabetic mice despite isotype-specific
differences in FcgammaR effector functions. J Immunol 2008;180(5):2863-2875.
498. Zhang
J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency
of interleukin 2-responsive T cells specific for myelin basic protein and
proteolipid protein in peripheral blood and cerebrospinal fluid of patients
with multiple sclerosis. J Exp Med 1994;179(3):973-984.
499. Quiniou-Debrie
MC, Debray-Sachs M, Dardenne M, Czernichow P, Assan R, Bach JF. Anti-islet
cellular and humoral immunity, T-cell subsets, and thymic function in type I
diabetes. diab 1985;34(4):373-379.
500. Naik
RG, Beckers C, Wentwoord R et al. Precursor frequencies of T-cells reactive to
insulin in recent onset type 1 diabetes mellitus. J Autoimmun 2004;23(1):55-61.
501. Roep
BO, Atkinson MA, Van Endert PM, Gottlieb PA, Wilson SB, Sachs JA. Autoreactive
T cell Responses in Insulin-dependent (Type 1) Diabetes Mellitus. Report of the
First International Workshop for Standardization of T cell assays. J Autoimmun
1999;13(2):267-282.
502. Schloot
NC, Meierhoff G, Karlsson FM et al. Comparison of cytokine ELISpot assay
formats for the detection of islet antigen autoreactive T cells. Report of the
third immunology of diabetes society T-cell workshop. J Autoimmun
2003;21(4):365-376.
503. Stratmann
T, Martin-Orozco N, Mallet-Designe V et al. Susceptible MHC alleles, not
background genes, select an autoimmune T cell reactivity. J Clin Invest
2003;112(6):902-914.
504. You
S, Chen C, Lee WH et al. Detection and characterization of T cells specific for
BDC2.5 T cell-stimulating peptides. J Immunol 2003;170(8):4011-4020.
505. Jang
MH, Seth NP, Wucherpfennig KW. Ex vivo analysis of thymic CD4 T cells in
nonobese diabetic mice with tetramers generated from I-A(g7)/class
II-associated invariant chain peptide precursors. J Immunol
2003;171(8):4175-4186.
506. Liu
CP, Jiang K, Wu CH, Lee WH, Lin WJ. Detection of glutamic acid
decarboxylase-activated T cells with I-Ag7 tetramers. Proc Natl Acad Sci U S A
2000;97(26):14596-14601.
507. Mallone
R, Nepom GT. MHC Class II tetramers and the pursuit of antigen-specific T
cells: define, deviate, delete. Clin Immunol 2004;110(3):232-242.